
Abstract
Identifying critically ill newborns who will benefit from whole genome sequencing (WGS) is difficult and time-consuming due to complex eligibility criteria and evolving clinical features. The Mendelian Phenotype Search Engine (MPSE) automates the prioritization of neonatal intensive care unit (NICU) patients for WGS. Using clinical data from 2885 NICU patients, we evaluated the utility of different machine learning (ML) classifiers, clinical natural language processing (CNLP) tools, and types of Electronic Health Record (EHR) data to identify sick newborns with genetic diseases. Our results show that MPSE can identify children most likely to benefit from WGS within the first 48 h after NICU admission, a critical window for maximally impactful care. Moreover, MPSE provided stable, robust means to identify these children using many combinations of classifiers, CNLP tools, and input data types—meaning MPSE can be used by diverse health systems despite differences in EHR contents and IT support.
Similar content being viewed by others


Introduction
Each year ~7 million infants worldwide are born with genetic disorders. Many are diagnosed and treated in the neonatal intensive care unit (NICU)1. Rapid progression of disease in acutely ill infants necessitates equally rapid diagnosis to implement personalized interventions. In recent years, whole genome sequencing (WGS) has emerged as a primary diagnostic tool2,3,4. An estimated one-fifth of NICU admissions involve Mendelian diseases, with WGS diagnostic yield commonly in the range of 25–50%5,6,7,8. However, identifying infants for WGS is difficult and time-consuming due to complex eligibility criteria, lack of neonatologist familiarity with WGS ordering, and evolving clinical features.
Manual review and prioritized selection of patient phenotypes is a time-consuming and expensive process, hindering WGS application in the NICU9,10. Since interpretation is phenotype-driven, incomplete or erroneous phenotype selection can result in false negative results. Failure to adhere to payer eligibility criteria can lead to refusal of reimbursement. Complicating this are the complexity of eligibility criteria and differences between payers. Recent efforts explore clinical natural language processing (CNLP) to automatically generate Human Phenotype Ontology (HPO)-based phenotype descriptions from clinical notes and have demonstrated diagnostic rates comparable to manual methods9,11. Automation promises scalability and efficiency in patient triage for sequencing. In previously published work, we have shown that combining CNLP with a machine learning-based prioritization tool, the Mendelian Phenotype Search Engine (MPSE), provides effective means to prioritize patients for WGS using electronic health records (EHRs)12,13.
Perhaps the greatest benefit from tools like MPSE will be seen by resource-limited healthcare systems which may lack the expertise, funding, or data necessary to develop in-house computational frameworks for genomics-based clinical care. Generalizability and adaptability are therefore essential. With these facts in mind, we have explored MPSE’s performance across multiple patient populations, data sources, and input data types. Our results reveal that MPSE is fast, flexible, generalizable, and highly portable.
Time to WGS order is also of critical importance in the NICU clinical setting. Earlier identification of patients likely to benefit from WGS, ideally as soon as possible after NICU admission, can significantly enhance care by enabling earlier disease diagnosis and timelier, more personalized interventions2,14,15. Once sequencing candidates have been nominated, the turnaround time from blood sample collection to diagnosis is typically multiple days or weeks14,16 but can occur in under 24 h9,17 using the most rapid protocols. As clinical sequencing turnaround times continue to decrease, the time from admission to nomination increasingly becomes the primary bottleneck in delivering timely genomic diagnoses. Recent studies on clinical WGS in the NICU have reported highly variable times-to-nomination, ranging from as early as 1 h after admission to over 100 days2,13,16. Here we show that MPSE can identify those children most likely to benefit from WGS within the first 24 h of admission to the NICU, a critical window for maximally impactful care. Moreover, we find that the MPSE scores of patients who are ultimately diagnosed with Mendelian diseases are higher than those of sequenced but non-diagnostic cases, a statistically significant trend that appears at 48 h post-admission and continues across the entire duration of the NICU stay. These findings argue for MPSE’s use as a proactive monitoring tool throughout the NICU stay.
Results
Comparing clinical NLP tool outputs
All phenotype data used in our initial publications12,13 was generated from patient clinic notes using the CNLP software CLiX. CLiX is a proprietary clinical NLP technology developed by the commercial healthcare analytics company Clinithink18. Given the ultimate goal of developing MPSE for adoption by diverse hospital and clinic systems, we sought to determine MPSE’s performance using phenotype data produced by other tools as well. We conducted a series of analyses on 5 different CNLP tools to compare their relative utility for use with MPSE: ClinPhen19, CLiX, cTAKES20, MedLEE21, and MetaMapLite22. A brief description of these tools is given in Supplementary Table 1. Before assessing MPSE’s performance using phenotype data produced by these different CNLP tools, we first compared the phenotype descriptions (HPO term sets) generated by these tools using the same sets of clinic notes—in this case, the notes from 1838 University of Utah NICU admits.
Term counts
Summary statistics for unique HPO term counts generated by each CNLP tool as well as the “manual” term sets identified by expert physicians are given in Supplementary Table 2. The HPO term sets used throughout this work were pre-processed by removing parent terms to keep only the most specific phenotype terms. In every case, the CNLP tools all produced larger HPO term sets per patient than did expert review. Among HPO term sets for the University of Utah NeoSeq patients, MedLEE yielded the fewest terms (average 31.4 terms per patient) while CLiX yielded the most terms (average 111.2 terms per patient), nearly twice as many as the next most prolific tool cTAKES (70.5 terms per patient). Unsequenced University of Utah NICU patients had significantly fewer terms in their phenotype descriptions than NeoSeq patients, consistent with our observations from other patient cohorts12.
Semantic similarity
We also calculated pairwise semantic similarity coefficients across all the NeoSeq phenotype sets. Semantic similarity is different from strict identity-based similarity measures, such as unweighted Jaccard similarity, in that two terms can be non-identical but still contribute positively to the similarity coefficient if they are neighbors, i.e., they lie near one another in the HPO directed acyclic graph. To estimate the probability that the semantic similarities of the term sets produced by the tools are statistically different from a null or random distribution, for each pairwise comparison, we sampled the HPO to yield two random term sets with sizes identical to the original sets. Semantic similarity coefficients were then calculated for these randomized sets and plotted alongside the real data in Supplementary Fig. 1. The pronounced separation between semantic similarity distributions of real and simulated data suggests the HPO term sets produced by the tools reflects a common underlying phenotypic reality that is being identified to a greater or lesser degree by all the tools. Consistent with this, the difference in mean similarity between real and simulated datasets is highly statistically significant for every tool by Student’s paired T-test, even after multiple test corrections (data not shown).
NLP sensitivity and accuracy
Next, we performed an orthogonal test of CNLP tool sensitivity and accuracy by calculating the overlap between the CNLP-generated phenotype sets and two distinct “ground truth” reference sets: expert-generated phenotypes and OMIM disease-associated phenotypes. The expert reference sets constituted the HPO term lists manually curated by physicians for the 65 sequenced University of Utah NICU patients23. The OMIM disease reference sets were restricted to the HPO phenotype terms associated with the OMIM disease diagnosis for the subset of 26 WGS-diagnosed Utah NICU patients. For these analyses, a CNLP term is considered a “true positive” if it or any of its parent terms are found in the ground truth set. This approach is justified by the ontological relationship between parent and child terms in HPO, where a child term inherently implies the presence of its parent term(s). Since HPO is structured in a manner where more specific terms (i.e., child terms) represent refined phenotypic descriptions, they subsume the more general terms (i.e., parent terms). For example, having Thrombocytosis (HP:0001894) necessarily indicates the presence of the parent phenotype Abnormal platelet count (HP:0011873).
Supplementary Fig. 2 shows sensitivity and accuracy distributions for CNLP terms sets compared with physician manual terms and OMIM disease-associated terms. The relative sensitivity among the CNLP tools roughly correlates with the tools’ average term counts (see Supplementary Table 2), which isn’t surprising; the more terms a tool generates, the more likely it will capture terms in the reference set. CLiX had the highest average sensitivity among the tools (manual terms sensitivity: 63%; OMIM terms sensitivity: 21%), while MetaMapLite had the lowest (manual terms sensitivity: 15%; OMIM terms sensitivity: 9%). The accuracy measure differs from sensitivity by controlling for the variable sizes of the CNLP term sets. Thus, a CNLP tool with high sensitivity may have a low relative accuracy if it has many more terms than another tool with lower sensitivity. ClinPhen had the highest average accuracy among the tools (manual terms sensitivity: 13%; OMIM terms sensitivity: 11%), while MetaMapLite had the lowest (manual terms sensitivity: 2%; OMIM terms sensitivity: 4%). Despite the modest sensitivity and accuracy of these CNLP tools compared to the “ground truth” sets, MPSE and other phenotype-driven clinical diagnostics tools such as GEM11, appear to be very robust against “noisy” phenotype data inputs (see next results for justification).
MPSE flexibly handles input data from a variety of sources
To further evaluate the practical utility of the MPSE algorithm, we conducted a comparative analysis of different CNLP tools and data types as inputs to MPSE. This analysis addresses several critical considerations for the deployment of MPSE in diverse clinical environments. First, we explored the interoperability of MPSE by assessing whether a model trained with data from one CNLP tool could reliably predict outcomes using data generated by a different tool. Second, we investigated the feasibility of using non-phenotype data with MPSE. Given the ultimate goal of developing MPSE for adoption by diverse hospital and clinic systems, the overarching purpose of these analyses is to better understand MPSE’s flexibility, robustness, and broader applicability in real-world clinical settings.
MPSE performance using different CNLP tools
To determine whether MPSE, trained with data from one CNLP tool, can reliably predict outcomes using data generated by a different CNLP tool, we began with the original MPSE model trained using CLiX-generated phenotype data from the RCHSD cohort. This model was used to make predictions on external phenotype data from the Utah cohort generated with ClinPhen, CLiX, cTAKES, MedLEE, and MetaMapLite. MPSE’s precision and diagnostic yield among top-scoring probands is plotted in panel A of Fig. 1. Apart from MetaMapLite, all the CNLP tools’ outputs work well when used as inputs for MPSE, a fact made clear by the high recovery rates of sequenced and diagnosed patients compared to choosing patients randomly for WGS. If MPSE was used to automatically select a volume of NICU patients for sequencing identical in size to the Utah NeoSeq study (n = 65) from among the 1838 total patients screened, CLiX and ClinPhen would maintain the NeoSeq study’s physician-mediated diagnostic yield (40%) throughout the top 50% of MPSE scores. This finding accords well with our previous publication, which showed high projected diagnostic yields from MPSE prioritization12.

Panels A, B display MPSE precision rates of patients manually selected for WGS (Panel A) and diagnostic yield for the subset of cases diagnosed by WGS (Panel B) using different CNLP tools. A CLiX-trained MPSE model from the RCHSD cohort was applied to phenotype data from 1838 University of Utah NICU patients generated by five different CNLP tools. Panels C, D display precision and diagnostic yield using MPSE models trained on four alternative data types (diagnosis codes, lab tests, medications, and all orders), compared to the corresponding HPO-based (CLiX) model trained on the same Utah cohort. A solid black reference line in each panel represents the precision or diagnostic yield expected from a model that chooses candidates at random, while the black dashed line in the diagnostic yield graphs (Panels B, D) indicates the NeoSeq study’s 40% total diagnostic yield. Figure generated with R ggplot2 software.
In addition to assessing yield among top-scoring probands, we also calculated cohort-wide performance metrics (see Supplementary Table 3). After the “native” CLiX data, ClinPhen yields the next-best overall predictions, with an average 20.7-point difference in MPSE score between cases and controls (p = 2.2e−14), the highest area under the ROC curve (AUROC = 0.91), and the highest area under the PRC curve (AUPRC = 0.45). MetaMapLite exhibited the lowest performance, likely caused by the relative dissimilarity between CLiX and MetaMapLite as seen in the low semantic similarity coefficients of CLiX-MetaMapLite term sets (Supplementary Fig. 1).
MPSE performance using alternative data types
To determine whether non-phenotype data types could be used with MPSE, we tested MPSE models built using diagnosis codes, lab tests, and medications and compared their predictive ability to phenotype-based models. A short description of these data types is given in Supplementary Table 4, and summary statistics of observation counts for each alternative data type are shown in Supplementary Table 5. MPSE models trained with alternative data types recovered fewer sequenced cases and diagnostic cases (Fig. 1B) among top-scoring probands than a corresponding phenotype-based model but still performed much better than a random model. Among the alternative data types, the ICD-10 based model yielded the best overall predictions, approaching the performance of the CLiX-based model (Supplementary Table 3). Our analysis suggests that non-phenotype structured data from patient EHRs is less effective than CNLP-derived phenotype data at identifying NICU sequencing candidates, but is a useful and valid substitute for CNLP phenotype descriptions if these are not available. However, it should be noted that this analysis was limited to only using the presence/absence of a lab test, medication order, etc. (see Methods) and not the test result or order specifications. An analysis using more precise laboratory and medication data may reveal higher MPSE performance when using these alternative data types.
Diagnostic performance using different CNLP tools
Lastly, we assayed the ultimate utility of the CNLP-derived phenotype term sets generated by each tool for clinical molecular diagnostic activities. For these analyses, we used an Artificial Intelligence (AI)-based gene prioritization tool called GEM11. Licensed from Fabric Genomics, by both RCHSD and the University of Utah, GEM is a commercial tool that combines HPO-based phenotype descriptions with WES and WGS sequences for rapid, AI-based diagnostic decision support. GEM was used by both RCHSD and the University of Utah for the original diagnosis of every sequenced proband in the datasets analyzed here.
Comparison of GEM’s previously published diagnostic performance to the prospective Utah data reported here provides a unique opportunity both to reexamine GEM’s performance using new, orthogonal data, and to assay the impact of using different CNLP tools on GEM’s diagnostic performance. These data are shown in Fig. 2. For reference, the original GEM benchmark results using manually curated HPO term sets for 119 RCHSD probands11 have been added for ease of comparison. Figure 2 shows the percentage of diagnosed Utah NeoSeq23 probands where the clinical molecular diagnostic genotype was reported by GEM among its top 1st, 2nd, 5th, and 10th gene candidates.

Bars show the proportion of diagnosed NeoSeq probands where the true causal genes were identified by GEM among the top 1st, 2nd, 5th, and 10th gene candidates. Each GEM run differed by the input HPO term lists, which were made by extracting phenotypes from patient clinical notes using CNLP (ClinPhen, CLiX, cTAKES, MedLEE, MetaMapLite) or manual physician review. The RCHSD benchmark (n = 119 patients) results from the original GEM paper are included for reference (redrawn with authors’ permission). Figure generated with R ggplot2 software.
Two major conclusions emerge from these data. First, diagnostic performance using CNLP-derived HPO data, regardless of the tool used to generate them, is nearly identical to the performance obtained using manual term sets, and second, the results shown here are highly concordant with the original published benchmarking results11. Moreover, the University of Utah data provides an entirely prospective orthogonal dataset, demonstrating transportability. These results demonstrate a powerful synergy between the MPSE approach and GEM. Namely, that the same HPO term sets used by MPSE for WGS prioritization can be directly consumed by GEM for downstream diagnoses. Moreover, Fig. 2 makes it clear that GEM can use HPO term sets, manually curated or CNLP derived, regardless of tool, without compromising downstream diagnostic accuracy.
MPSE can identify patients who would benefit from WGS within the first 48 h of NICU admission
Our initial work and that presented here has demonstrated MPSE’s ability to accurately identify sequencing candidates by aggregating information from the entirety of the patient’s NICU stay. These findings underscore the tool’s effectiveness in a retrospective context, using all notes up to the date at which the attending physicians place the order for WGS. However, the ultimate test of value lies in validating the real-time utility of MPSE. Early identification of patients who would benefit from WGS, as soon as possible after NICU admission, could significantly enhance care, enabling earlier disease diagnosis and more timely interventions2,14,15.
To measure MPSE’s real-time utility, we calculated daily MPSE scores for each patient in our Utah cohort using only HPO terms extracted with CLiX from clinical notes present in the EHR at 24-h intervals, beginning at the moment of their admission. Thus, each patient had a series of MPSE scores for each day spent in the NICU from admission to discharge. Longitudinal MPSE scores for patients who received a molecular diagnosis by WGS (diagnostic), those for whom WGS did not identify a molecular diagnosis (non-diagnostic), and patients who were not selected for WGS (unsequenced) are summarized in Supplementary Table 6 and plotted in Fig. 3 to help visualize the change in MPSE score over time among these groups. By the end of the first day (0–24 h) in the NICU, both diagnostic and non-diagnostic sequenced cases had statistically significantly higher MPSE scores than did those who were not selected for sequencing (unsequenced mean: −48.4; diagnostic mean: −32.1, p = 1.4e−5; non-diagnostic mean: −28.2, p = 9.3e−6). Additionally, diagnostic cases had significantly higher average MPSE scores than non-diagnostic sequenced cases beginning 48 h post-admission (non-diagnostic mean: −24.7; diagnostic mean: −9.0; p = 0.018) and continuing thereafter.

Panel A shows MPSE score distributions across the first 96 h in the NICU for diagnostic (red) and sequenced but non-diagnostic (blue) patients as well as unsequenced NICU patients (green). Diagnostic and non-diagnostic sequenced patients had significantly higher MPSE scores than unsequenced patients beginning 0–24 h after admission. Diagnostic patients had significantly higher MPSE scores than non-diagnostic patients beginning at 48 h post-admission. Boxplot comparison significance levels: *** (p < 1e−5); * (p < 0.05). Panel B shows MPSE score trajectories for these groups across the first 30 days in the NICU. Solid lines show the mean MPSE score per group and the shaded regions cover one standard deviation from each mean. Panel C shows the probability of at-risk patients being classified as a WGS candidate by MPSE (i.e., MPSE score >2 standard deviations above the unsequenced mean score) as a function of time. Cox proportional hazards regression analysis confirmed the significantly increased rate of MPSE candidate selection for diagnostic and non-diagnostic patients selected for WGS compared to unsequenced patients, with hazards ratios of 18.8 (95% CI 11.6–30.6; p = 1.9e−32) and 9.8 (95% CI 6.2–15.3; p = 2.8e−23) respectively. Diagnostic patients were also selected by MPSE at a higher rate than sequenced but non-diagnostic patients (HR = 2.0; 95% CI 1.1–3.9; p = 0.03). Figure generated with R ggplot2 software.
In addition to differences in MPSE scores between unsequenced, non-diagnostic, and diagnostic patients, there are also significant differences in the daily change in MPSE score (day-N delta) between these groups. Both diagnostic and non-diagnostic sequenced cases saw greater average day-N delta than unsequenced controls throughout the first 30 days post-admission (Supplementary Table 6). Importantly, the greatest difference in MPSE score increase was observed for the day-one delta, i.e. the change in MPSE score during the first 24 h post-admission, with average diagnostic MPSE score rising by 18.4 points, average non-diagnostic MPSE score rising by 7.7 points, and average unsequenced MPSE score rising by only 2.7 points. These day-one delta differences were statistically significant for each subgroup comparison (diagnostic vs unsequenced p-value = 0.0015; non-diagnostic vs unsequenced p-value = 0.004; diagnostic vs non-diagnostic p-value = 0.027).
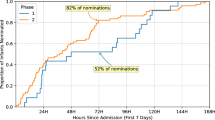
To gain further insight into the temporal dynamics of MPSE’s predictive capabilities across the NICU stay, we estimated the proportional risk of being identified as a WGS candidate by MPSE using Cox proportional hazards regression analysis24. The estimated probability and 95% CI of candidate selection by MPSE for diagnostic, non-diagnostic, and unsequenced patients is plotted in Panel C of Fig. 3. Using a rule-of-thumb MPSE score threshold (calculated individually for each day) of 2 standard deviations above the mean score of unsequenced control patients, diagnostic patients were flagged by MPSE at a significantly higher frequency and speed than both unsequenced patients (HR = 18.8, p = 1.9e−32) and non-diagnostic sequenced patients (HR = 2.0, p = 0.03). At 48 h post-admission, MPSE had already flagged 74% (17 of 23) of diagnostic patients, 47% (17 of 36) of non-diagnostic sequenced patients, and only 6% (100 of 1773) of unsequenced patients. Furthermore, by 9 days post-admission all diagnostic patients had been flagged by MPSE or were censored as a result of death or NICU discharge, highlighting the speed at which MPSE was able to determine correct clinical action for this group of patients. Supplementary Table 7 contains the daily score threshold, the number of candidates assessed by MPSE, and the cumulative number of patients who reached the score threshold as part of this longitudinal analysis.
Discussion
We previously demonstrated that an MPSE-based automated pipeline for prioritizing acutely ill infants for whole genome sequencing can meet or exceed diagnostic yields obtained by time-consuming manual review of clinical notes and histories12. Our work here using the clinical histories of 2941 NICU admits drawn from two different institutions serves to expand on those original findings. A supplemental benchmark of MPSE’s predictive performance using various statistical classifiers revealed Naive Bayes and Support Vector Machines are robust techniques for phenotype-drive patient prioritization (see performance benchmark in Supplementary Table 8). As such, we have provided pre-trained models for both techniques in the public MPSE GitHub repository (https://github.com/Yandell-Lab/MPSE). Assessing MPSE’s flexibility with handling phenotype data from different CNLP tools showed that MPSE’s performance is largely agnostic with respect to upstream CNLP tool. Moreover, we found that structured EHR data, such as ICD diagnosis codes, can provide an effective alternative for prioritizing patients for WGS in health settings where access to clinical notes and NLP pipelines is problematic. These two features of MPSE combine to greatly lower the IT burden for deployment.
Our longitudinal analyses demonstrate that MPSE can identify those children most likely to benefit from WGS within the first 48 h of admission to the NICU, a critical window for maximally impactful care, and in some cases long before human case review led to the same conclusion. Moreover, the consistent performance of MPSE over the first 30 days post-admission argues for its utility as a monitoring tool throughout the entirety of a patient’s NICU stay. These findings make clear MPSE’s potential value as a real-time clinical support tool and how MPSE could be used to improve cost savings2,3,14,15 and the timeliness and effectiveness of care.
Finally, we have also shown that the same HPO term sets used by MPSE for prioritization for WGS, regardless of the CNLP tool generating them, can be directly consumed by the AI tool GEM for downstream molecular diagnoses, further speeding and facilitating personalized care. These results collectively demonstrate that MPSE provides fast, flexible, and highly scalable means for prioritizing critically ill newborns for whole genome sequencing.
The American College of Medical Genetics and Genomics (ACMG) 2021 Practice Guideline recommends clinical genome sequencing as a first or second-tier test for infants with one or more congenital anomalies25. We compared the efficiency of using this ACMG guideline to using MPSE for automated patient selection. Presence of one or more HPO terms corresponding to common and clinically impactful congenital anomalies was used to approximate the ACMG selection criteria (Supplementary Table 9). The MPSE diagnostic yield at 48 h post-admission was 13% (17 diagnostic patients out of 134 selected). The ACMG guideline-based diagnostic yield was 4% (13 diagnostic patients out of 321 selected). These results suggest MPSE can provide a 3.3-fold enrichment in diagnostic yield compared to ACMG criteria alone while achieving essentially the same number (17 vs. 13) of diagnosed children.
Despite overwhelming clinical evidence that NICU and PICU genome sequencing saves lives and reduces costs3,14,15,25, several barriers still hinder its broad adoption. One significant obstacle is reimbursement for testing by payers26,27,28. Broader, more inclusive eligibility criteria simplify the candidate selection process, but increase cost-burden and decrease diagnostic yield. This can result in payer hesitancy to reimburse WGS, especially for negative results. While more stringent selection criteria can decrease cost-burden and increase diagnostic rates, they also increase the time-burden of candidate assessment. Collectively, our results demonstrate MPSE’s potential to standardize, speed, and render more scalable the WGS selection process. A recently published clinical implementation study of MPSE provides additional support for this conclusion13. Moving forward, we will further evaluate the utility of MPSE as part of the recently funded GeneKids project whose goal is to bring genome-based healthcare to the USA Intermountain West, and explore secondary applications of MPSE in assessing eligibility for reimbursement29.
Methods
Datasets
Our clinical cohort comprised 293 probands who underwent rWGS at Rady Children’s Hospital in San Diego (RCHSD), 85 of whom received a molecular diagnosis for a Mendelian disorder. These 293 individuals were selected from symptomatic children enrolled in prior studies exploring the diagnostic rate, time to diagnosis, clinical utility, outcomes, and healthcare utilization of rWGS between July 26, 2016, and September 25, 2018, at RCHSD (ClinicalTrials.gov identifiers: NCT03211039, NCT02917460, and NCT03385876). All participants presented with symptomatic illnesses of unknown etiology and suspected genetic disorders. The diagnosed cases provide a real-world population displaying various Mendelian conditions arising from diverse modes of disease inheritance and disease-causing genotypes. An additional 756 patients admitted to the NICU at RCHSD in 2018 were also included. These patients were added to enrich the dataset with a broader spectrum of phenotypes not necessarily associated with Mendelian diseases. In total, the RCHSD dataset used in this study contains a total of 1049 individuals. Additional details are provided in refs. 2,7,9,14,15.
We also employed a second, independent dataset consisting of 1838 newborn patients admitted to the University of Utah level III NICU from January 2020 to December 2022, the approximate study period of the Utah NeoSeq Project. The Utah NeoSeq Project was a multidisciplinary, longitudinal rapid genome sequencing program conducted at the University of Utah to improve genetic diagnosis in critically ill infants in the NICU23. Within the Utah cohort of 1838 patients, 65 were selected for rWGS based on manual chart review as part of the NeoSeq study. 26 of these children received a molecular diagnosis.
The need for Institutional Review Board approval at Rady Children’s Hospital for this study was waived as all patient data used in this project was previously collected as part of studies approved by the Institutional Review Boards of Rady Children’s Hospital. The University of Utah Institutional Review Board approved the use of human subjects for this research under a waiver for the requirement to obtain informed consent. None of the results reported in this manuscript can be used to identify individual patients. This research was conducted in full compliance with all relevant ethical regulations, including the Declaration of Helsinki.
Statistical classifiers
Various Machine Learning (ML) classifiers were used to train multiple independent MPSE models. The original MPSE algorithm employed a Naive Bayes classifier and is described in detail in our proof-of-concept work12. Additional MPSE models were trained using the following ML classifiers: K-Nearest Neighbors (KNN)30, Decision Trees (DT)31, Random Forests (RF)32, Logistic Regression (LR)33, Gradient Boost Machine (GBM)34, Support Vector Machine (SVM)35, and Multi-Layer Perceptron (MLP)36. Each classifier was implemented using scikit-learn, a general-purpose machine learning library written in the Python programming language37. Each method was run with scikit-learn version 1.4.2 default parameters. Models were trained using the RCHSD cohort (n = 1049) and internally validated using stratified K-fold cross validation (K = 8). Each of the trained classifiers was subsequently validated externally with five randomly split subsets of the Utah cohort (n = 419 per set). These results are described in Supplementary Table 8 and its associated text.
Phenotype and alternative data types
Highly curated, manually created HPO-based phenotype descriptions were provided for each of the 65 sequenced University of Utah probands. CNLP-derived phenotype descriptions were generated for all 1049 RCHSD probands and the 1838 University of Utah probands by CNLP analysis of clinical notes recorded during NICU stay using CLiX18. Additional CNLP-derived phenotype descriptions were generated using the following text mining and CNLP methods: ClinPhen19, cTAKES20, MedLEE21, and MetaMapLite22 for the University of Utah probands. CLiX and ClinPhen generate HPO terms directly from clinic notes, while cTAKES, MedLEE, and MetaMapLite return Unified Medical Language System (UMLS)38 Concept Unique Identifiers (CUIs) which were then mapped to HPO terms using the UMLS Metathesaurus’ network hierarchy. For sequenced patients, clinical notes dated post-WGS were excluded from analysis to remove notes containing sequencing results. In addition to CNLP-derived phenotype data, ICD-10 diagnosis codes, laboratory tests, medications, and other hospital orders were collected for all University of Utah probands to serve as alternative data sources for MPSE modeling. Unlike free-text clinic notes, these alternative data types are stored in structured form within the University of Utah’s Enterprise Data Warehouse (EDW) and were extracted using automated database queries. Laboratory tests and medication orders were encoded as binary variables to represent the presence/absence of a test or order without the context of the test result or medication dosage, frequency, adherence, etc.
Calculating semantic similarity between phenotype sets
We calculated pairwise semantic similarity between physician- and CNLP-generated HPO term sets for patients using the Python package PyHPO v3.1.4. PyHPO calculates the similarity between two HPO sets as described in refs. 39,40. We also generated simulated, ‘randomized’, HPO term sets for every proband in order to provide a “null” distribution for our semantic similarity calculations. For each “real” pairwise set comparison, simulated HPO sets of equal size were randomly sampled from the HPO. Semantic similarity between these sets of randomly sampled terms was then calculated using the same method that was used to compare the manual and CNLP sets to one another. Significant differences between “real” and “randomized” set similarities were tested for using paired Student’s T-test.
Testing the precision and diagnostic yield of MPSE
We calculated the precision and diagnostic yield at K for the 5, 10, 25, 45, and 65 top-scoring patients by dividing the number of true positives by K. For calculating precision, a true positive was defined as a WGS-selected patient that was classified (flagged) by MPSE as a WGS candidate based upon the contents of its clinical notes using a particular C-NLP tool, i.e., its MPSE score >0. For calculating diagnostic yield, a true positive was defined as a WGS-diagnosed patient that was flagged by MPSE (MPSE score >0).
Gene prioritization using NLP-derived phenotype descriptions
Artificial Intelligence (AI)-based prioritization and scoring of candidate disease genes for the diagnosed probands was performed using Fabric GEM11. GEM is a commercial tool for AI-assisted clinical interpretation of WES and WGS. It has been licensed by the University of Utah and Rady Children’s Hospitals from Fabric Genomics Inc. Additional licensing information is available from Fabric Genomics Inc. GEM inputs are genetic variant calls in VCF format and case metadata, including parental affection status, and patient phenotypes in the form of HPO terms. For these analyses, GEM was run six times for each NeoSeq proband, varying only the input HPO lists, the first with the proband’s physician-selected terms and the remaining runs using HPO term sets created with the five different CNLP tools.
Longitudinal analysis of MPSE scores
We conducted a longitudinal analysis of MPSE scores across each University of Utah patient’s NICU stay. For this analysis, we employed the original MPSE model trained on the RCHSD cohort and utilized CLiX-derived HPO phenotype descriptions. Human Phenotype Ontology (HPO) terms were timestamped according to the date of the clinical note, with each day’s cumulative HPO list being used for MPSE score calculations. Six sequenced University of Utah patients had to be excluded from this analysis because a data upload error left clinic note timestamps unavailable for these patients, leaving 59 remaining sequenced patients available for longitudinal analysis. Subsequently, we calculated average daily MPSE scores for unsequenced controls, sequenced but not diagnostic cases, and diagnostic cases. We estimated the daily probability MPSE would recommend patients from these groups for sequencing using a score threshold (calculated individually for each day) of 2 standard deviations above the mean score of unsequenced control patients and calculated the associated hazards ratio with Cox proportional hazards regression analysis using the R survival package (v3.7.0).
Comparing diagnostic yield between MPSE and ACMG practice guideline
We calculated the 48 h post-admission diagnostic yield achieved by MPSE versus that achieved using American College of Medical Genetics and Genomics (ACMG) criteria. University of Utah cohort patients (n = 1838) were selected for WGS by MPSE at 48 h post-admission using the score thresholding approach detailed in the longitudinal analysis section (above). Patients from the same group and time point were again selected for WGS according to the 2021 ACMG Practice Guideline25 for the use of exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. This guideline recommends clinical genome sequencing as a first or second-tier test for patients with congenital anomalies prior to one year of age. Thus, the selection criteria for ACMG was one or more HPO terms indicating a congenital anomaly. Because phenotype terms representing congenital anomalies are scattered throughout the HPO hierarchy and not related under a single top-level parent term, we manually gathered a list of 40 HPO terms corresponding to common and clinically impactful congenital anomalies. See Supplementary Table 9. The proportion of selected individuals who were previously diagnosed by WGS was then calculated using these two methods.
Data availability
The datasets used in this study are available from the corresponding author upon reasonable request.
Code availability
MPSE source code, documentation, and synthetic datasets are available on GitHub (https://github.com/Yandell-Lab/MPSE).
References
Christianson, A., Howson, C. P. & Modell, B. March of Dimes. https://onprem.marchofdimes.org/materials/global-report-on-birth-defects-the-hidden-toll-of--WAk00l8AFdheR3Hyz4TRcSVNc5tvO5T0ziR4-AlnXYs.pdf (2006).
Farnaes, L. et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom. Med. 3, 10 (2018).
Dimmock, D. et al. Project baby bear: rapid precision care incorporating rWGS in 5 California children’s hospitals demonstrates improved clinical outcomes and reduced costs of care. Am. J. Hum. Genet. 108, 1231–1238 (2021).
Kingsmore, S. F. & Cole, F. S. The role of genome sequencing in the NICU. Annu. Rev. Genom. Hum. Genet. 23, 427 (2022).
French, C. E. et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 45, 627–636 (2019).
Petrikin, J. E. et al. The NSIGHT1-randomized controlled trial: rapid whole-genome sequencing for accelerated etiologic diagnosis in critically ill infants. NPJ Genom. Med. 3, 6 (2018).
Kingsmore, S. F. et al. A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am. J. Hum. Genet. 105, 719–733 (2019).
Clark, M. M. et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 3, 16 (2018).
Clark, M. M. et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci. Transl. Med. 11, eaat6177 (2019).
James, K. N. et al. Partially automated whole-genome sequencing reanalysis of previously undiagnosed pediatric patients can efficiently yield new diagnoses. Npj Genom. Med. 5, 1–8 (2020).
De La Vega, F. M. et al. Artificial intelligence enables comprehensive genome interpretation and nomination of candidate diagnoses for rare genetic diseases. Genome Med. 13, 153 (2021).
Peterson, B. et al. Automated prioritization of sick newborns for whole genome sequencing using clinical natural language processing and machine learning. Genome Med. 15, 18 (2023).
Juarez, E. F. et al. A machine learning decision support tool optimizes WGS utilization in a neonatal intensive care unit. Npj Digit. Med. 8, 72 (2025).
Dimmock, D. P. et al. An RCT of rapid genomic sequencing among seriously ill infants results in high clinical utility, changes in management, and low perceived harm. Am. J. Hum. Genet. 107, 942–952 (2020).
Sweeney, N. M. et al. Rapid whole genome sequencing impacts care and resource utilization in infants with congenital heart disease. NPJ Genom. Med. 6, 29 (2021).
The NICUSeq Study Group Effect of whole-genome sequencing on the clinical management of acutely ill infants with suspected genetic disease: a randomized clinical trial. JAMA Pediatr. 175, 1218–1226 (2021).
Owen, M. J. et al. An automated 13.5 h system for scalable diagnosis and acute management guidance for genetic diseases. Nat. Commun. 13, 4057 (2022).
Clinithink. Clinithink: AI solutions company, clinical data solutions for life science & healthcare. https://www.clinithink.com (2025).
Deisseroth, C. A. et al. ClinPhen extracts and prioritizes patient phenotypes directly from medical records to expedite genetic disease diagnosis. Genet. Med. 21, 1585–1593 (2019).
Savova, G. K. et al. Mayo clinical Text Analysis and Knowledge Extraction System (cTAKES): architecture, component evaluation and applications. J. Am. Med. Inform. Assoc. JAMIA 17, 507–513 (2010).
Friedman, C., Shagina, L., Lussier, Y. & Hripcsak, G. Automated encoding of clinical documents based on natural language processing. J. Am. Med. Inform. Assoc. JAMIA 11, 392–402 (2004).
Demner-Fushman, D., Rogers, W. J. & Aronson, A. R. MetaMap Lite: an evaluation of a new Java implementation of MetaMap. J. Am. Med. Inform. Assoc. JAMIA 24, 841–844 (2017).
Malone Jenkins, S. et al. The Utah NeoSeq Project: a collaborative multidisciplinary program to facilitate genomic diagnostics in the neonatal intensive care unit. Npj Genom. Med. 10, 1–11 (2025).
Cox, D. R. Regression models and life-tables. J. R. Stat. Soc. Ser. B Methodol. 34, 187–202 (1972).
Manickam, K. et al. Exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability: an evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 23, 2029–2037 (2021).
Reuter, C. M. et al. Yield of whole exome sequencing in undiagnosed patients facing insurance coverage barriers to genetic testing. J. Genet. Couns. 28, 1107–1118 (2019).
Phillips, K. A. et al. US private payers’ perspectives on insurance coverage for genome sequencing versus exome sequencing: a study by the Clinical Sequencing Evidence-Generating Research Consortium (CSER). Genet. Med. J. Am. Coll. Med. Genet. 24, 238 (2021).
Grant, P. et al. Out-of-pocket and private pay in clinical genetic testing: a scoping review. Clin. Genet. 100, 504–521 (2021).
Friesen, S. New collaboration aims to provide genetic diagnoses for thousands of kids. https://uofuhealth.utah.edu/newsroom/news/2024/08/new-collaboration-aims-provide-genetic-diagnoses-thousands-of-kids (University of Utah Health Newsroom, 2024).
Cover, T. & Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 13, 21–27 (1967).
Azar, A. T. & El-Metwally, S. M. Decision tree classifiers for automated medical diagnosis. Neural Comput. Appl. 23, 2387–2403 (2013).
Kulkarni, V. Y. & Sinha, D. P. K. Random Forest Classifiers: A Survey and Future Research Directions. Int. J. Adv. Comput. 36, 1144–1153 (2013).
Dreiseitl, S. & Ohno-Machado, L. Logistic regression and artificial neural network classification models: a methodology review. J. Biomed. Inform. 35, 352–359 (2002).
Bentéjac, C., Csörgő, A. & Martínez-Muñoz, G. A comparative analysis of gradient boosting algorithms. Artif. Intell. Rev. 54, 1937–1967 (2021).
Cortes, C. & Vapnik, V. Support-vector networks. Mach. Learn. 20, 273–297 (1995).
Schmidt, B. & Hildebrandt, A. Deep learning in next-generation sequencing. Drug Discov. Today 26, 173–180 (2021).
Pedregosa, F. et al. Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Humphreys, B. L. & Lindberg, D. A. The UMLS project: making the conceptual connection between users and the information they need. Bull. Med. Libr. Assoc. 81, 170–177 (1993).
Robinson, P. N. et al. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 83, 610–615 (2008).
Pesquita, C. et al. Metrics for GO based protein semantic similarity: a systematic evaluation. BMC Bioinforma. 9, S4 (2008).
Acknowledgements
The preparation of this manuscript was supported by the University of Utah’s National Library of Medicine training grant (T15LM007124, PI - Eilbeck), NIH grant UL1TR002550 from NCATS to E.J. Topol (with sub-award to Rady Children's Institute for Genomic Medicine), and the Warren Alpert Foundation. The Utah NeoSeq Project was funded by the Center for Genomic Medicine at the University of Utah Health, ARUP Laboratories, the Ben B. and Iris M. Margolis Foundation, the R. Harold Burton Foundation, and the Mark Miller Foundation. This work utilized resources and support from the Center for High Performance Computing at the University of Utah. The computational resources used were partially funded by the NIH Shared Instrumentation grant 1S10OD021644-01A1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Library of Medicine or the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
M.Y., B.P., and S.F.K. wrote the manuscript. B.P., M.Y., B.M., E.J.H., and S.F.K. designed the study. M.Y., B.P., and E.J.H., developed the MPSE algorithm. B.P., E.J.H., B.M., and J.L. performed the data analysis. B.P. generated all figures. E.F.J, S.M.J., and M.T.F. compiled cases and clinical evidence. E.F. and M.N.B. provided feedback on features and development. Y.L., M.G.R., M.N.B., S.F.K, and S.M.J. sponsored the project and provided helpful discussions and edits of the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.Y. is a co-founder and consultant for Fabric Genomics Inc. and has received consulting fees and stock grants from Fabric Genomics Inc. M.G.R. is a shareholder of Fabric Genomics Inc. E.F. is an employee of Fabric Genomics Inc. B.M. and E.J.H. have received consulting fees and hold equity from Fabric Genomics Inc. S.F.K. is an associate editor for npj Genomic Medicine but was not part of the peer review or decision making of this manuscript. The remaining authors declare that they have no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Peterson, B., Juarez, E.F., Moore, B. et al. MPSE identifies newborns for whole genome sequencing within 48 h of NICU admission. npj Genom. Med. 10, 47 (2025). https://doi.org/10.1038/s41525-025-00506-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-025-00506-3
