
Abstract
The surge in commercial and civilian spaceflight enables the systematic and longitudinal, large-scale biospecimen collection to understand the prospective effects of space travel on human health. The Genomics and Space Medicine (Space Omics) project at BCM-HGSC involves a comprehensive biospecimen collection plan from commercial/private space flight participants. The manuscript addresses the critical gaps in the biospecimen collection process including details of the informed consent process, a provision for subjects to obtain custom CLIA-WGS reports, a data dictionary and a LIMS enabled biobank. The manuscript also discusses the biospecimens collection, processing methodologies and nucleic acid suitability for Omics data generation. Results from Axiom-2 mission where, 339 biospecimens were collected using ‘Genomic Evaluation of Space Travel and Research (GENESTAR)’ manual, at two different sites, showed that 98% of the blood samples and 91.6% of the non-blood samples passed the QC requirements for Omics assays, underscoring the reliability and effectiveness of the GENESTAR manual.
Similar content being viewed by others

Introduction
As humanity looks towards long-duration missions and habitation in space, understanding the biological implications of space travel is crucial. Unlike ground studies, where rigorous scientific studies can be conducted by involving statistically significant sample cohorts, biospecimens available to study the effects of space are highly underpowered. As of December 2024, only 714 people have traveled into space (defined higher than 50 miles (80 km) above mean sea level, according to the U.S. Armed Forces, and National Aeronautics and Space Administration (NASA)1. Earlier biospecimen collections were driven in partnership with NASA’s Human Research Program (HRP) studies2. More recently, through the eXploration Platforms and Analog Definition (EXPAND) program, the Translational Research Institute for Space Health (TRISH) implemented proactive strategies in program management, processes, and infrastructure3. These efforts will help enhance engagement with space flight providers, commercial launch providers, space biomedical researchers, and government agencies with the goal of making data-informed decisions.
Biological and physiological data from subjects have been collected and analyzed to assess the impact of space flights4,5,6,7. However, the details of the underlying molecular consequences of space flight are poorly understood,2. This includes Immune dysregulation and the latent virus reactivation8, Oxidative stress mechanisms contributing to DNA damage and aging as well as pathways of bone loss and muscle atrophy9. Also unknown are the molecular mechanisms of Spaceflight-Associated Neuro-ocular Syndrome (SANS), and Frequent headaches during missions10,11. Changes in the gut microbiome and host-pathogen dynamics during spaceflight also require deeper exploration12. Jugular vein thrombosis (JVT) is a rare condition caused by blood clots in the jugular vein and a known threat for astronauts also without details of molecular etiology13,14. Having the biological data will be informative in analyzing rapid and subtle changes in the human body. A comparative assessment of various bio-fluids, including whole blood, plasma, urine, body swabs, stool and saliva, obtained from the subjects can reveal specific biomarkers, was previously reviewed15,16.
Advances in Omics technologies such as genomics, transcriptomics, proteomics, and metabolomics have had a significant impact on human genetic research, cancer detection and treatment, and personalized medicine. Therefore, there is growing interest in the application of Omics technologies to study the effects of microgravity, radiation exposure, microbiome, and other environmental factors on individuals experiencing either short- or long-duration commercial space flight. The resulting data can be valuable for advancing terrestrial health care as well17. Omics technologies were applied to analyze the data from the NASA Twin study and, more recently, Inspiration4, the first all-civilian orbital spaceflight mission12,18,19. Sequencing was also used to analyze Clonal Hematopoiesis of Indeterminate potential mutations in a retrospective specimen cohort20. A proof-of-concept study showed that exosome sequencing could be applied to study the pathophysiology of Space Associated Neuro-Ocular Syndrome (SANS) in subjects21. However, space health studies remain vastly underpowered. The challenges of space health research are analogous to those faced in terrestrial rare disease research, where the small number of affected individuals (often fewer than 1 in 10,000) leads to similar issues with underpowered studies. Achieving sufficient statistical power will require scaling up participation, collecting biospecimens at multiple time points, generation of data from multi-omics assays, leveraging data from multiple missions, terrestrial analogs, and fostering global collaboration22. The recent surge in commercial space missions provide a unique opportunity to systematically collect biospecimens from crew using a standardized methodology for both immediate analysis and biobanking, thus helping to achieve the required statistical power. Also, there is a critical gap in knowledge and know-how of how national and international participants can be enrolled in such Omics studies and how clinical-grade reports detailing pathogenic variants and pharmacogenomics markers can be provided to them.
The Genomics and Space Medicine project (‘Space Omics’) at the Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) was initiated in August 2022, and is designed to study pre-, in-, and post-flight biological specimens using an array of Omics assays, including clinical Whole Genome Sequencing (WGS), research assays (RNA-Seq, microbiome, proteomics among others), and biobanking for future use to gain insights into the impact of space travel. Biospecimens from up to 40 individuals will be collected over a five-year period making this the largest Omics dataset and biobank in this field. The knowledge gained from the collected biospecimens and the Omics data from diverse subjects on these commercial missions will be crucial for future mission planning and to enhance human health in space and on Earth.
Multiple sites across the country will be collecting biospecimens using the standardized set of protocols presented here as a ‘Genomic Evaluation of Space Travel and Research (GENESTAR) manual for biobanking and Omics data generation. To ensure practical relevance and feasibility, the GENESTAR manual was developed by actively incorporating feedback, suggestions, and constraints from key commercial space partners. The GENESTAR manual was developed at Baylor College of Medicine (BCM), Houston, Texas, and subsequently implemented at the University of Central Florida, Florida, to handle collections at Kennedy Space Center (KCS), Florida, for both the Axiom-2 and Axiom-3 missions. A third site, CALTECH, Hawthorne, California was also recently added to handle biospecimen collections on the West Coast. Additionally, the University of Pennsylvania (U Penn) site handles biospecimen collections from centrifuge training at the NASTAR facility in Southampton, Pennsylvania, thereby broadening institutional participation and collaboration.
Biospecimen collections for the Axiom 2 private mission, which was a 10-day mission (May 21 – May 30) involving a four-person multinational crew to the International Space Station, is described here to demonstrate the methodology for specimen collection, processing, and biospecimen requirements. Additionally, 154 biospecimens from Axiom mission 3 (January 18, 2024 – February 9, 2024) have also been successfully collected using these protocols, and collections for a 3rd private space mission are underway.
Results
Standard operating procedures were developed for the collection of a wide range of biospecimens from each crew member (Fig. 1). These samples were aliquoted or harvested to obtain either nucleic acids or derivative samples to carry out specified Omics studies, such as genomic, transcriptomic, proteomic, microbiome, and single-cell sequencing assays (Table 1). The remaining aliquots deposited in a biobank for future use along with the generated Omics data and through proper permissions, are accessible to other researchers.

Collection details shown here are for each of the pre-and post-flight timepoints including one-time collection of blood for CLIA-WGS. For a typical mission, there will be 2–3 pre-flight and 2–3 post-flight collections.
Data dictionary based on Athena-Ohdsi
A data dictionary was created using the Athena-Ohdsi database to describe primary biospecimens and the Omics assays used in the GENESTAR manual23. This dictionary described the 13 primary biospecimens obtained from collections such as blood, body swabs, saliva, stool, and urine, and their derivatives (Table 2); and the six planned Omics assays (Table 2 and Table 3). For each of these 19 entries, the field name, concept ID, concept code, concept name, Athena Domain name have been provided (Table 2 and Table 3). The data dictionary along with the sample metadata from the biobank will be submitted to the TrialX EXPAND database that will host the Space Omics project data.
Informed consent process and personal data privacy
Participants are enrolled into the Commercial Astronaut Data Repository (CADRE) Institutional Review Board protocol that covers the participant enrollment and data governance (H-50346), MESH protocol (H-52035) that oversees ethical and privacy considerations, and TRISH Privacy Protocols that ensure participant anonymity and data security. The Space Omics project is an integral part of this framework. This enrollment is carried out at an informed consent briefing meeting where the EXPAND PI, study coordinators, clinical geneticist and subject experts participate. At this briefing, participants are taken through the study’s purpose, risks, and potential benefits, as well as how their genetic data will be used, stored, and shared. Navigating international laws, such as the EU’s GDPR or the US’s HIPAA Privacy Rule, and binding to the laws of the host country and space agency that a subject belongs to can pose unique challenges to this enrollment process. The informed consent process is tailored to ensure that participants understand the potential implications of their participation, including the possibility of increased public interest. A strong emphasis is placed on privacy and confidentiality protections.
The subjects can opt out of the Space Omics enrollment at any point and to address this, biospecimen destruction procedures are in place in the GENESTAR protocol. Each subject receives a coded ID which is also grouped with a mission ID to accommodate tracking of information for the repeat flyers. These IDs are shared for use in the GENESTAR protocol to collect biospecimens. Upon receiving the biospecimens at BCM-HGSC, these IDs go through a second round of coding (de-identification) to enter into the biobank and those are the IDs that are used to generate Omics data through all stages of the project and also data submission to the TrialX EXPAND database. These are the IDs and the associated data that will be released to the public (including this report) which helps to further blind the data from subjects as well as the private agency responsible for that mission (Axiom Space in this report).
Biobanking
A functional LIMS-enabled biobank to receive, store, and track Space Omics samples has been created. The Exemplar Laboratory Information Management System (LIMS) is built upon a Java 2 Platform, Enterprise Edition (J2EE) architecture and utilizes a Tomcat server for its operation. This LIMS has exceptional adaptability, offering extensive configuration options to align seamlessly with any laboratory workflow. It is designed to efficiently manage biospecimen tracking from initial intake, through library preparation and sequencing, to the final quality control stages. Moreover, this system is engineered for high throughput and minimal latency, featuring a user-friendly, code-free search functionality. Security is a paramount concern, with the system employing a comprehensive security framework and offering role-based access control to accommodate the varied roles within laboratory staff. Additionally, the Exemplar LIMS supports seamless integration capabilities with laboratory automation systems and the analysis pipeline, enhancing operational efficiency and data coherence across laboratory processes (Fig. 2).

LIMS-enabled biobank was set up at BCM-HGSC to receive, process, and to track samples for future use.
Upon arrival, barcodes on collected biospecimens are scanned into LIMS and marked as “received”. The biospecimen number and barcodes relative to the information in the biospecimen intake forms along with the other metadata are cross checked, and any discrepancies are noted. At this step a second round of de-identification of the subject IDs is done and then uploaded into LIMS. Any problems with the sample condition are also recorded. Samples are either directly stored at −80 °C or aliquoted into smaller vials for storage. In some instances, such as urine collected in a specimen container and whole blood collected in the CPT tubes, the primary biospecimens are processed to separate different components (PBMCs or urine cell pellet, etc). In such a scenario, new barcodes are created to label the tubes containing the aliquots and the derivative products, and that information is also entered into the LIMS (Fig. 2). Biospecimens are then advanced to different Omics pipelines and all major lab/informatic processes that the biospecimens undergo. Any aliquots made from that biospecimen are additionally barcoded and linked to the original biospecimen barcode. The chain of custody and biospecimen tracking in the LIMS are achieved by scanning the barcode on the biospecimen tube into LIMS before each processing step to generate Omics data. BCM-HGSC is the genomics data provider. Both the Omics data and Biospecimen metadata will be submitted to the EXPAND database created and managed by TrialX, a clinical research and space health informatics company also supported by TRISH. Requests for data and sample accesses will be managed by EXPAND Data Privacy and Release Board (DPRB).
Overview of biospecimens and collection time points
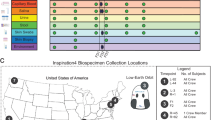
The type of samples selected for collection were chosen based on their minimally invasive, cost- and time-effective properties, easily accessible with the requirement of non-specialized equipment or expertise, and high return in data relevance to health assessment by variable Omics (Fig. 1). The collection times have been calibrated using the exact splashdown time (For Axiom-2, it was May 30, 2023, 11:04 PM EDT), where the first 24 h are designed as R + 0 followed by R + 1 (06.01.2023 9:00 AM CST/10:00 AM EDT), when the first samples were collected for this mission), and so on. The biospecimens presented here were collected from four Axiom-2 mission subjects across six collection time points over four months as detailed in Table 4. The biospecimens were collected at 90, 30, and 3 days pre-launch (L-90, L-30, and L-3), 1, 6, and 14 days after the return (R + 1, R + 6 and R + 14). Blood was collected for the CLIA-WGS from three subjects on R + 14 and from the fourth subject on R + 29 (Table 4). In total, for the Axiom-2 mission, 339 primary samples were collected from the four subjects, (Table 4). No inflight collections were performed. All but L-3 collections happened at Axiom Space Inc. headquarters in Houston, Texas. The biospecimens were shipped within ~2 h after collection for initial processing at the Human Genome Sequencing Center (HGSC) at Baylor College of Medicine (BCM), Houston, Texas. While for the L-3 timepoint, biospecimens were collected at Kennedy Space Center (KCS), Florida using the same SOP. The initial processing of those L-3 biospecimens was performed at Burnett School of Biomedical Sciences (BSBS), the University of Central Florida, Florida, and shipped back to BCM-HGSC at recommended shipping conditions as in the GENESTAR manual for further processing and biobanking.
Subject de-identification process
Space flight providers assign random initial de-identified subject IDs to the participants which the participants are aware of and these IDs are also used to label the biospecimens collection kits, which makes it easy for the participants to identify the kits and provide the respective biospecimens. HGSC assigns the second de-identified IDs to ensure compliance with blinding protocols and to prevent participants from knowing the details of their samples during analysis and publication, a secondary de-identification process was implemented. This additional step also ensures that their sample identity from others involved in the different steps in the workflow. The second de-identification of the subject IDs involved generating a 9-character alphanumeric code using a random code generation script. These codes will also have a prefix SO (for Space Omics). The IDs will not be used more than once even if the subject is a repeat flyer. These IDs will be assigned upon sample receipt at the time of biobank intake.
Biospecimens collections and their derivatives—blood samples
For research purposes, blood samples were collected via venipuncture from each crew member at six time points. One blood biospecimen was collected post-return to perform genome sequencing and generate clinical data.
The collection tubes were labeled with the subject ID, the collection number, analyte, and the aliquot number (Example: Subject ID-1-Saliva-01). Additionally, each tube was labeled with a unique barcode to ensure accurate and efficient identification of the samples.
Whole blood was collected at L-90, L-30, L-3, R + 1, R + 6 and R + 14. The R + 14 collection was performed on only two subjects for a total of 22 biospecimen.
Whole blood was collected in K2 EDTA tubes at six time points, L-90, L-30, L-3, R + 1, R + 6 and R + 14. Each collection involved three K2 EDTA tubes of 3 mL each. The R + 14 collection was for only two subjects, for a total of 62 EDTA blood biospecimen.
One tube of K2 EDTA from each subject and collection was used to isolate plasma that was then aliquoted into four 300 µL aliquots. For the Axiom-2 mission this added up to 88 plasma aliquots.
The second K2 EDTA tube was used for DNA extraction. To avoid batch effect, the DNA from the blood collected at different time points was obtained through a single isolation. The total DNA yield varied between 26.38 µg and 92.0 µg and the DNA yield per mL of blood was between 13.1 µg/mL and 46 µg/mL, respectively (Fig. 3). The 260/280 ratio of ≥1.8 indicated no major protein contamination. The DNA blood isolation from astronaut SO5QL3KG6H resulted in higher yield per mL of blood, followed by SO6NJ3UF7T over the different collection times. The average yield per mL of blood over collection time was 32 µg: SO6NJ3UF7T, 40 µg: SO5QL3KG6H, 23 µg: SO8KF3EZ1C, 21 µg: SO9WB8CN8A. The third K2 EDTA tube and the plasma aliquots were stored as-is at −80 °C for future use.

DNA was isolated from one of the blood samples collected in K2 EDTA tubes (n = 22) and PAXgene DNA tubes (n = 4) to perform whole genome sequencing and CLIA-WGS, respectively. The yield per ml blood and ratio 260/280 are depicted on Y-axis with n = 26.
Whole blood for the purpose of generating CLIA-WGS data was collected from all four subjects, with each subject providing blood in 2.5 mL in PAXgene DNA tubes at R + 6 or R + 29 (Table 4). Total DNA yields varied between 45.08 µg and 89.76 µg, while the DNA yields per milliliter of blood were between 22.54 µg/mL and 44.88 µg/mL. The yield per mL of blood collected the same day showed minimal variation between the PAXgene and the EDTA collection tubes. This DNA was utilized to generate CLIA-WGS data and was analyzed for reportable genetic variants in a specific set of genes.
For each collection, 4 mL of blood was collected in Cell Preparation Tubes (CPT) from which Peripheral Blood Mononuclear Cells (PBMCs) were isolated. The final volume of PBMCs harvested was between 1.5 and 3 mL among the subjects. PBMC counts and viability for subject SO6NJ3UF7T from L-30 and L-3 was negligible and failed at this QC step. For the remaining 10 biospecimens, cell viability was >80% and the cell count was between 800 and 1600 cells/µL (Fig. 4). The total number of PBMCs harvested among these 10 samples was between 1.3 and 3.2 million, while the minimum required number of cells for single-cell sequencing is only 1000 cells.

PBMCs were isolated from two pre-launch and one post return blood collections to generate single cell RNA-seq data. Cells were counted manually using a hemocytometer. PBMC counts/µl and the total number of the cells used for sequencing were depicted on the y-axis, a line is drawn at 1000 cells across all samples to indicate a minimum requirement for sequencing.
Between 4–6 mL of plasma was isolated from the 22 blood samples collected in 10 mL BCT tubes and the plasma was stored at −80 °C until cfDNA extractions. Cell-free DNA was extracted using the entire volume of plasma obtained. The DNA yields and fragment sizes when evaluated using the High Sensitivity DNA Bioanalyzer kit showed the yields between 0.65 and 12.8 ng/mL of plasma (Fig. 5). Typical cfDNA size profiles of mononucleosome cfDNA (range: 75 and 250 bp, with a166bp peak); a portion of di- (range: 300–400 bp), and tri-nucleosomes (range: 450–650 bp) representation was also seen.

CfDNA was isolated from the plasma samples obtained from the blood samples collected in blood BCT tubes (n = 22) to perform cfDNA sequencing. The yields per ml of plasma are depicted on Y-axis.
RNA was isolated from the 22 blood samples collected in 2.5 mL RNA PAXgene tubes (Table 4). RNA yield was between 0.58 µg and 2.55 µg per mL of blood (Fig. 4). The RIN scores of these samples were between 7.1 and 9.4 and DV200% values between 84% and 94% (Fig. 6). As noticed earlier, the astronaut SO5QL3KG6H had a higher average RNA yield (1.90 µg per mL of blood) than the others.

RNA integrity (RIN and DV200%) was assessed using Agilent Bioanalyzer on nanochips. The total RNA yields, RIN and the DV200% values are depicted on the y-axes, with n = 4 for each time point except for R + 14 collection, where biospecimens were available for only two subjects.
In summary, a total of 102 blood samples which includes 98 samples for research purposes: K2 EDTA DNA (22), RNA (22) EDTA plasma (22) and BCT Plasma (22) and 10/12 PMBC biospecimens and four samples of DNA in PAXgene tubes for CLIA-WGS, with passing QC metrics for Omics data generation, indicating a 98% success rate.
Biospecimens collections and their derivatives- non-blood samples
Body swab, saliva, stool, and urine samples were collected from each crew member across four time points: L-90, L-30, L-3, and R + 6. 16S rRNA sequencing data was generated for samples collected at L-30, L-3, and R + 6 time points. The 16S sequence reads were mapped against the SILVA Database version 138, which contains sequences from the v4 region of the 16S rRNA gene. Nucleic acids were not extracted from L-90 biospecimens. A total of 160 body swab samples from 10 different body sites were collected (Supplementary Fig. 1). DNA was extracted using 400 µl from 120 out of 160 body swab samples, excluding the L-90 biospecimens. Successful sequencing libraries were obtained from 119 samples with library yields between 98.3 and 1000 ng. UMB sample from subject SO5QL3KG6H failed at library preparation. The 16S gene sequencing yielded mapped reads between 18 and 145,071 reads. The rarefaction cutoff was set at 562 mapped reads and 107 samples passed this metric. Out of the 12 samples that did not meet this cut-off, one was saliva and 11 were swabs from different body sites and from L-30 and R + 6 timepoints. Five of these 11 samples were from subject SO8KF3EZ1C, four from SO9WB8CN8A and one each from the other two subjects. Sixteen urine biospecimens between 90–105 mL were collected in a specimen container from which, supernatant and cell pellets were harvested and stored as aliquots at −80 °C. Supernatant aliquot volumes are between 30–35 mL while the cell pellets aliquots are 250 µl each.
Successful 16S rRNA libraries were generated from all 12 urine samples from which DNA was extracted, with library yields ranging between 126.0 and 826.0 ng. All the urine samples have generated mapped reads ranging from 5,930 – 145,071 mapped reads. A total of 15 saliva samples were collected. The biospecimen volumes varied from 2 mL to 6.8 mL and were divided into 250 µL aliquots. One saliva aliquot from each biospecimen was used for DNA extraction. Successful 16S rRNA libraries were generated for all the samples with library yields ranging between 111.1 and 492.9 ng. The 16S gene sequencing yielded mapped reads from each saliva sample between 305 and 31,847 reads. The biospecimen collected at L-3 from subject SO8KF3EZ1C failed the rarefaction cut-off as it generated only 305 mapped reads. A total of 16 stool samples were collected and the DNA extractions were performed on 200 µl of the sample. Bacterial 16S rRNA sequencing was performed using 2 µl of the extracted DNA from the samples, without quantification. Libraries were successfully generated from all the 12 samples, excluding the L-90 samples. A minimal variation in the library yields (range of 3.01- 3.5 µg) was observed among these samples. The 16S gene sequencing yielded mapped reads between 61,820 and 87,796 mapped reads.
16S rRNA data as a QC
Bacterial 16S rRNA gene sequencing data from the non-blood samples is presented here to document biospecimens quality and the efficiency of sequencing technology employed. Out of 155 biospecimens across four analytes selected for 16S rRNA sequencing, one failed at the initial quality control step and did not proceed to library preparation. Additionally, 12 samples did not produce sufficient mapped reads and were considered failures. The sequencing data were successfully generated for the remaining 142 biospecimens, representing a 91.61% success rate. Overall, the microbiome data was similar across the four analytes, the subjects and time points studied for observed operational taxonomical units (OUTs) and for Shannon alpha diversity (Fig. 7) confirming the usability of the collected biospecimen. The mean number of observed OTUs showed slight variation with a median of 35 and the Shannon diversity index indicated a more or less even spread of microbial abundance among all the subjects and time points. Microbiota did not change significantly among the subjects, across the time points, with adjusted p-values in the range of 0.5–0.9 for richness and 0.8–0.9 for Shannon diversity.

Box plots depict high-level alpha diversity metrics across four biospecimen types including urine, stool, body swabs, and saliva. The observed Operational Taxonomical Units (OTUs) and Shannon alpha diversity provide a visual representation of the microbial diversity, highlighting the consistency in the biospecimens collected, processed and 16S microbiome data generated from them.
Discussion
There has been a growing interest in recent years both from government and private industry to explore space including space tourism, returning humans to the moon, and planning missions to Mars24. It is therefore important to understand the effects of space travel on human health. The collected biospecimens allow for the identification of trends in molecular and physiological changes. However, we acknowledge that the inherently small cohort size in spaceflight studies poses challenges to achieving full statistical power for all goals. To address this, the GENESTAR protocol emphasizes standardized biospecimen collection, biobanking, and data sharing to enable future meta-analyses and collaborative studies that could achieve sufficient power to answer more specific research questions.
Collection of biospecimens and generation of Omics data are essential to understand the genetic, physiological, cellular, and microbiome changes related to space travel. Rigorous Standard Operating Procedures (SOPs) for specimen collection have been developed along with a LIMS-enabled biobank at HGSC that tracks specimen details and storage information. This document, called Genomic Evaluation of Space Travel and Research (GENESTAR) incorporated biospecimen collection procedures tailored to support all current and anticipated biological and genomic measurements.
In GENESTAR, we have standardized the complete set of protocols, based on our knowledge of biospecimens requirements for genomics and other molecular assays, as well as those adopted from earlier studies, NASA-Human Research Program core measures25 and Space Omics and Medical Atlas (SOMA)18 as described recently for the Inspiration4 mission. For the first time in the GENESTAR manual, a data dictionary, a LIMS-enabled biobank, and a customized provision for subjects to obtain CLIA-WGS are provided. Coupled with the informed consent process, GENESTAR fills these critical gaps that were not addressed by those previous studies.
Here, we present our collection efforts based on biospecimens gathered from the Axiom 2 mission, representing a typical commercial crew mission from a subject cohort that spent 10 days in microgravity. Several of the presented biospecimen collection methods provide a validation of the recently reported methods, which were intentionally synchronized to allow for joint biospecimens collections by us and the SOMA procedures18 for a private mission. Further, our group has exclusively collected samples from Axiom-2 and Axiom-3 missions, in total from six individuals, which will allow for easy comparison of results across different missions.
As the physiological changes are very dynamic during a standard spaceflight mission, obtaining Omics measures across multiple time points before and after the travel to space is important for scientific significance. Pre-flight biospecimen collections help assess and establish a baseline and biospecimen collections shortly after the return, help assess the recovery process and any long-term health implications. A collection of biospecimens from at least 2 to 3 pre-flight and 2 to 3 post-flight time points is recommended so that any outliers in data can be identified, then either ignored or accounted for. Given the typical short duration (under 30 days) of these commercial/private missions, there is a narrow window within which we must collect the samples. Thus, we were limited to six biospecimen collections within a four-month window. The six-time points include L-90, L-30, L-3, R + 1, R + 6, and R + 14, which can capture the effects of space travel by comparing the biospecimen collected from pre and post-flight, along with an unambiguous baseline. In that context, Axiom 2 missions’ biospecimen collection time points align with the 3 days inspiration 4 mission collection time points of L-92, L-44, L-3, R + 1 and R + 45 and R + 8218. In contrast, the NASA twin study was a one-year mission with a wider window with time points including L-180, L-120, L-60, launch, five inflight collections, R0, R + 60, R + 120, and R + 180, albeit overlaps with the above missions12. Further, the GENESTAR manual is adaptable if there is an option for additional time points to be included. The primary biospecimens collected include blood, body swabs, saliva, stool and urine samples, which have provided secondary products such as plasma, urine cell pellet and supernatant. These sample types were selected after carefully reviewing the sample types that were collected during the two previous multi-omics studies12,18. This multi-tiered approach in biospecimens collection allows for a comprehensive multi-omics analysis. Blood, urine, stool, swabs, and saliva provide cost-effective, non-invasive, and easy sampling options and therefore inherently qualify as appropriate biospecimen types to obtain Omics data from subjects 16.
We are additionally offering the option of a CLIA-WGS for the first time to the participants26. The HGSC launched a CAP/CLIA certified clinical laboratory in 2016 (CLIA# 45D2027450) and therefore can deliver clinical genetic testing data and interpretation, enabling the return of clinical results reports to individuals, via their clinical caregivers. Through CLIA-WGS, the subjects in the Space-Omics program may elect to receive results for reportable genetic variants in 205 genes, including American College of Medical Genetics and Genomics (ACMG) 59 gene set recommended for secondary findings reporting27. A ‘Space Omics’ clinical-grade report will be returned to the participants unless the participants specifically opt-out of the results return.
Both our research and NASA HRP aim to advance our understanding of human space exploration. However, our research specializes in genomics and multi-omics studies, which offer a molecular perspective and integrate diverse Omics data types to elucidate the effects of spaceflight on human biology.
A data dictionary using the Athena-Ohdsi database components to describe biospecimen and Omics assay types is described. This will facilitate efficient information dissemination to other users globally, for data analysis and interpretation across various missions23.
A versatile LIMS-enabled biobank to store and track Space Omics samples has been developed. All biospecimens and their derivatives when stored in the biobank are barcoded and tracked properly. LIMS at BCM-HGSC also provides support for tracking of samples as they progress through the different Omics pipelines as well as to track and retrieve the generated data. Overall, the established biobank enhances the efficiency, accuracy, and security of biospecimen management which are essential for a robust biobank to function28.
In the presented study, the extracted DNA and RNA underwent quality assessment and were determined to be of sufficient quality and quantity to conduct multiple genomics assays listed in Table 1. A proteomics assay run on plasma samples is particularly valued for its ability to simultaneously provide highly sensitive and specific measurements of multiple proteins from small biospecimen volumes29. As plasma contains various molecules such as proteins, metabolites, nucleic acids, and lipids, its analysis may reveal exceptionally specific biomarkers associated with space travel30,31. Plasma will be used to generate high-throughput proteomics datasets, such as Olink Explore HT. Cell-free DNA is a potential biomarker for diagnosis and prognosis in conditions such as cancer, cardiovascular and neurological diseases32. Further, cfDNA can be used to measure physiological stresses during space missions33. The PBMCs derived from blood samples were of acceptable quality and quantity, allowing for the generation of single-cell RNA sequence data (Manuscript under preparation). DNA extracted from both low biomass samples including body swabs, saliva, and urine, and high biomass biospecimen including stool was utilized for 16S rRNA gene sequencing. The sequencing methods were adapted from those developed for the NIH-Human Microbiome Project and the Earth Microbiome Project34. 16S rRNA gene sequencing was favored over microbiome WGS for cost reasons and the extracted nucleic acids are stored for future use. A total of 155 samples consisting of 12 urine, 12 stool, 120 body swabs, and 11 saliva samples were submitted for bacterial microbiome sequencing. Successful sequence data was obtained for 142 out of 155 samples (91.6%). Twelve samples were body swab samples out of 13 failed samples. Out of the four subjects, SO8KF3EZ1C had the most samples that failed followed by SO9WB8CN8A. This outcome can be attributed to various factors, including but not limited to disparities in the biospecimen collection times among different individuals, contact ___location, and in the case of body swabs, the pressure applied during the collection process18,35.
Nevertheless, microbiome sequencing generated 91.6% of usable data underscoring the validation of the collection methodology, efficiency and quality of both the sampling and sequencing processes. As a further confirmation of this data’s usability, Alpha diversity and richness measurements were performed to QC the microbial diversity (Fig. 7).
GENESTAR contributes to identifying biological responses to space travel, such as changes in the microbiome, transcriptomics, and cfDNA. While this study provides crucial insights, it is not powered for interventional assessments but rather for identifying targets that could inform future countermeasure studies, as well as the identification of gaps in research.
These biospecimens are being collected at different locations across the country using the GENESTAR manual and shipped to HGSC after initial processing. The unpredictability of shuttle schedules can cause sample collection delays, making it difficult to collect biospecimens as planned, especially if collections happen during late hours and weekends. Shipping samples from collection sites to the local processing labs and back to BCM-HGSC in Houston for long-term storage can be challenged by transport and weather-related interruptions and must be documented thoroughly for any deviations. GPS trackers are included in the sample shipments and extra dry ice is packed to avoid running into problems due to such interruptions.
Further enhancements to the GENESTAR manual are also planned. For instance, the in-flight whole blood collections are still not part of the manual. Traditionally, blood in-flight is collected using venipuncture. However, there are several microdevices that allow easy self-collection of blood for diagnostic and research purposes. These microdevices can collect several hundred microliters of whole blood, enough to isolate plasma/serum to perform certain Omics assays like proteomics and metabolomics. Our group is evaluating them for use in the GENESTAR protocol and, if found effective in microgravity, will be used for in-flight sample collections. The GENESTAR manual will also be used to collect biospecimens from age and gender matched controls to isolate noise in the data and conduct ground-based analog experiments such as centrifugation training.
The GENESTAR protocol was designed to prioritize operational convenience, ensure continuity in biospecimen collection across missions, and seamlessly integrate into the schedules of astronauts and private space organizations. All the three multi-omics studies undertaken so far GENESTAR, SOMA18 and NASA-HRP12 captures the core biospecimens so that the cohort of samples and data available to study the effects of space on human health are as inclusive as possible12,19,25 while there are assays that GENESTAR and SOMA are not generating the exact datasets (16S for GENESTAR vs metagenomic WGS for SOMA; targeted proteomics vs untargeted proteomics) or that the GENESTAR is not generating lipidomics data immediately, the biospecimens collected and biobanked are available for later data generation from these assays. For the first time, in the GENESTAR manual, a data dictionary, a LIMS-enabled biobank, and a customized provision for subjects to obtain CLIA WGS among the three protocols. In summary, the methodology presented lays a strong foundation for future studies and allows for the comparison of existing Omics datasets.
Methods
Data dictionary based on Athena-Ohdsi
A detailed data dictionary for each of the primary biospecimens collected has been developed using the Athena-Ohdsi database to standardize biospecimen types and medical terminology, aligning codes with the Observational Medical Outcomes Partnership (OMOP) standards, which are a set of guidelines and tools designed to standardize the collection, formatting, and analysis of observational healthcare data. OHDSI Standardized Vocabularies comprise over 10 million concepts from 136 vocabularies13. Athena is a comprehensive vocabulary repository and searchable database within OHDSI.
Biospecimens collections and their derivatives—blood samples
Venipuncture was performed in the antecubital fossa after sanitizing that area and blood was drawn using a butterfly needle (BD Biosciences, Cat#367281) and a lure lock vacutainer adapter (McKesson, Cat#364815/458363). The blood collection volumes involved, 3 mL of blood in each of the three K2 EDTA blood collection tubes (VWR, Cat#367856), 4 mL of blood in one CPT cell preparation tube (CPT, BD Biosciences, Cat#362760), 10 mL of blood in one Streck cell-free DNA BCT tube (cfDNA BCT, Streck, Cat#230470) and 2.5 mL of blood in one PAXgene blood RNA tube (RNA PAXgene, Fisher, Cat#762165), for a total of 25.5 mL collected from each subject at each time point. It is not feasible to collect 25.5 mL volumes of blood multiple times on the same day, as there are limitations on the amount of blood that can be collected from the astronauts and they also might be participating in other studies requiring blood samples.
An additional 2.5 mL of blood was collected from each subject in CAP CLIA validated PAXgene DNA tubes (VWR, cat# 10589-986) during the second collection after the return, to be used exclusively for CLIA-WGS data generation. Samples collected locally in Houston were immediately transported to the Human Genome Sequencing Center at Baylor College of Medicine for processing. Blood samples collected in Florida at the Kennedy Space Center (KCS), were transported to the University of Central Florida, within 2 h for initial processing, which included preparation of PBMCs and plasma isolation, as discussed below.
Processing of blood samples
Whole blood was collected at six time points from the four subjects at L-90, L-30, L-3, R + 0, R + 6 and from 2 subjects at the R + 14 timepoint.
Whole blood in BD Hemogard K2 EDTA tubes was collected from each subject in three tubes of 3 mL each per time-point. DNA was isolated from one tube (see section Nucleic Acid Extractions from blood samples), plasma from the second tube and the third tube was stored at –80 °C as received.
Plasma was isolated from one of the K2 EDTA blood samples collected from each subject at each time point. The blood was centrifuged at 1,000 g for 10 min at room temperature. The plasma was collected and aliquots of 300 µL were distributed into four matrix tubes (ThermoFisher, Cat#3741-WP1D-BR) of 300 µL to avoid additional freeze-thaw cycles. The aliquots were stored at −80 °C.
Blood (2.5 mL) was collected in PAXgene tubes for CLIA-WGS at R + 6 from 3 subjects and at R + 29 from the fourth subject and tubes were stored at −80 °C. DNA was isolated using a CAP-CLIA validated protocol (see below section Nucleic Acid Extractions from blood samples).
Blood samples were collected in CPT tubes for PBMC isolation from each subject and at each time point. The blood samples were mixed immediately by gently inverting the CPT tube eight to 10 times before centrifuging at 1,800 g for 20 min at room temperature. Half of the plasma was aspirated without disturbing the cell layer and the cells were transferred to a 15 mL conical tube. 2% FBS (Thermo Fisher, cat# 26140079) in PBS (Gibco, cat#10010-023) was added to resuspend the cells and the volume was brought to 15 mL. The cells were mixed gently by inverting five times and then centrifuged at 120 g with break-off for 10 min. The supernatant was aspirated and discarded without disturbing the cell pellet. The pellet was dislodged by tapping the tube with index finger, resuspended in 2% FBS in PBS and the volume was brought to 10 mL. The pellet in FBS-PBS was mixed gently by inverting five times followed by a spin at 120 g for 10 min. The supernatant was discarded, and the pellet was resuspended in 1 mL of RPMI (Cytiva, cat#SH30027.01) medium. An equal volume of freezing medium [30% DMSO (MP Biomedicals, cat#194818) 40% FBS (Thermo Fisher, cat# 26140079), and 30% RPMI (Cytiva, cat#SH30027.01,)] was added to the cell suspension. The cell suspension was then transferred into two cryovials, 1 mL each. The cells were cryopreserved using an isopropanol freezing container and then placed in −80 °C freezer overnight. These cryovials were stored in liquid nitrogen until use.
To count the cells, the cell suspension (50 µL) was mixed with trypan blue (50 µL) (Sigma-Aldrich Cat. No. 72-57-1) at a 1:1 ratio and 10 µL of the mixture was loaded onto the hemocytometer (Bulldog Bio, DHC-N420). Then, the hemocytometer was placed on the microscope stage (Zeiss Axio Vert. A1), optimally focused, and manually counted to determine the total, live, and dead cell density and viability.
Blood samples were collected in cell-free DNA (cfDNA) BCT tube (Streck, Cat#230470) at a volume of 10 mL. Each biospecimen was centrifuged at 1600 g for 10 min at room temperature. Plasma was separated from each sample, transferred into 15 mL conical tube and stored at −80 °C until cfDNA extractions. The cfDNA extractions were performed as described below.
Blood samples were collected in PAXgene blood RNA tube (Fisher, Cat#23-021-01) at a volume of 2.5 mL from each subject. The RNA was isolated as described below.
Nucleic Acid Extractions from blood samples
Genomic DNA was extracted from K2 EDTA tubes and PAXgene blood collection DNA tubes on the Chemagic Prime 8 using the Chemagic Prime DNA blood kit (Revvity, cat# CMG-1497) according to the manufacturer’s instructions in a CAP-CLIA certified laboratory. DNA quality and quantity were assessed by electrophoresis and fluorescent nucleic acid stain (PicoGreen, Thermo Fisher).
Cell-free DNA was extracted from the BCT tube using the Apostle MiniMax High-efficiency cfDNA Isolation kit (VWR, cat# 76409-757), as per the manufacturer’s instructions. The quantification and fragment size profiling of cfDNA was determined using a 2100 bioanalyzer (Agilent Inc., Santa Clara, CA, USA) with the Agilent High-Sensitivity DNA kit. (Part #5067-4626, Agilent) and the cfDNA was stored at −20 °C until further use.
RNA was extracted from the PAXgene blood collection RNA tubes on the Chemagic Prime 8 using the Chemagic Prime total RNA blood 4k kit (Revvity, cat# CMG-1484) according to the manufacturer’s protocol. The RNA was utilized for total RNA sequencing and the remaining RNA was stored at −80 °C for future use. RNA quality (RIN and DV200) and quantity were assessed by Agilent Bioanalyzer.
Biospecimens collections and their derivatives- Non-blood samples
Body swab, saliva, stool and urine samples were collected from each crew member across four time points: L-90, L-30, L-3 and R + 6 and were proceeded as below until nucleic acids were extracted.
The ten body swab collections involved eight wet and two dry swabs. The wet body swab locations included post-auricular (EAR), axillary vault (PIT), volar forearm (ARM), occiput (NAP), umbilicus (UMB), gluteal crease (GLU), glabella (TZO), and toe web space (WEB) while the two dry body swab collections included oral (ORC) and nasal (NAC) (Supplementary Fig. 1 and Supplementary Table 1). For wet swab collections, the swab (Isohelix, Cat#MS-02) was moistened in nuclease-free water and rubbed back and forth on the skin for about 25–30 times along the surface, applying firm pressure while rotating the swab head continuously. The swab was then placed into the matrix tube provided (ThermoFisher, Cat#3741-WP1D-BR) with 400 µL of DNA/RNA shield (Zymo Research, Cat#R1100-50). The biospecimen collection process was repeated for all seven other wet swab locations. Buccal swab collection was performed by inserting the dry swab into the mouth and rubbing firmly against the inside of the cheek or underneath the lower or upper lip for about a minute. The nasal swabs were collected by inserting the entire soft tip of the dry swab into one nostril until resistance was experienced. The swab was rubbed in a circle around the nostril four times for at least 15 s. The process was repeated in the other nostril. Swabs were returned to the RNA/DNA shield (Zymo Research, Cat#R1100-50) as mentioned above. All the swabs were stored at −80 °C until nucleic acid extractions were performed.
Crude saliva was collected from each subject into an OMNIgene ORAL (DNAgenotek, Cat# OME-505) saliva collection tube that contains a solution for collection, stabilization, storage, and transportation of the sample. Each subject was allowed to spit repeatedly into the saliva collection tube until the “fill to” line, which is equivalent to 3 mL. The saliva was mixed by inverting the tube. 250 µL aliquots were distributed into cryovials under sterile conditions and the aliquots were stored at −80 °C.
Stool biospecimen collections were carried out in DNA Genotek OMNIgene Gut (OMR 205) tubes using the accessory and spatula provided in the kit. Typically, 545 mg of stool is collected and mixed with the stabilizing buffer supplied in the tube for a total of 4 mL. The samples were stored at −80 °C until nucleic acid extractions were performed.
Approximately, 90 mL of the urine sample from each subject were collected in a sterile Samco Bio-Tite Specimen Container (SAMCO, Cat#13-711-65) to which 6.3 mL of urine conditioning buffer (Zymo research, Cat# D3061-1-140) is added immediately and mixed thoroughly. From each container, three aliquots of 30 mL each were prepared in 50 mL conical tubes. The samples were centrifuged at 3,000 g for 15 min at room temperature to obtain a cell pellet and supernatant. The supernatant was transferred into clean 50 mL conical tubes and stored at –80 °C while the urine cell pellet was resuspended in 250 µL of DNA/RNA shield (Zymo Research, Cat#R1100-50) and stored at −80 °C until nucleic acid extraction.
Nucleic Acid Extractions from non-blood samples
Nucleic acid extractions were performed on low biomass (body swabs, saliva, and urine), and high biomass (stool) samples collected from each subject across three time points (L-30, L-3 and R + 6), excluding the L-90 biospecimens.
DNA was extracted from an aliquot of urine cell pellet (250 µL) and skin swab biospecimen (400 µL) types using the Zymobiocs (96) Kit (Cat# 27500-4-EP) following the manufacturer’s protocol. Each extraction was eluted into 50 µL of nuclease-free water (Invitrogen, Ref# AM9906, Lot# 2211042) and the samples were ready for immediate use or frozen until needed.
DNA was extracted from 200 µl of primary stool and a 250 µl- saliva aliquot using the Qiagen PowerMicrobiome kit (Cat# D4303) following the manufacturer’s protocol. The VIAFLO96 pipetting station (Integra, Part# 6001) was used for these extractions, and samples were eluted in 100 µl of nuclease free water. The samples were ready for immediate use or frozen until needed.
16S rRNA gene sequencing as a QC metric for non-blood samples
16S rRNA gene sequencing was chosen for microbiome analysis rather than whole genome sequencing (WGS) due to budget constraints. 16S rRNA gene sequencing provides a cost-effective and sufficient method for profiling microbial diversity and community structure, the project objectives.
The 16S rRNA gene sequencing methods were adapted from those developed for the NIH-Human Microbiome Project and the Earth Microbiome Project14. Briefly, the 16S rRNA v4 region was amplified by PCR using primers that contain adapters for sequencing on the Illumina platform. Additionally, single-index barcodes were incorporated into the reverse primer allowing PCR products to be pooled and sequenced directly. The PCR template volume was 5 µL for urine and skin swab biospecimen types and 2 µL for saliva and stool biospecimen types. Platinum Taq (Invitrogen – Cat# 15966005) was used for amplification.
Library QC was performed using a combination of gel electrophoresis (1% agarose) and an automated PicoGreen assay (Invitrogen Quant-iT P7589) using the Hamilton STARlet Liquid Handler. Finalized libraries were normalized and pooled followed by loading on the Illumina MiSeq platform using the 2x250bp (16S V4) paired-end protocol. The 16S sequence reads were mapped against the SILVA Database version 138 which contains sequences from the v4 region of the 16S rRNA gene. Data is accessible through TrialX EPAND database.
Human subjects research
All subjects consented and biospecimens were collected and processed under the approval of the Institutional Review Board at Baylor College of Medicine, under MESH protocol H-52035.
Manuscript preparation
Figure 1 was created with BioRender.com under the Baylor College of Medicine Institutional license.
Data availability
The data are accessible through TrialX EXPAND database.
References
Astronaut/Cosmonaut Statistics Available from https://www.worldspaceflight.com/bios/stats.php.
Alwood, J. S. et al. From the bench to exploration medicine: NASA life sciences translational research for human exploration and habitation missions. NPJ Microgravity 3, 5 (2017).
Urquieta, E. et al. Establishment of an open biomedical database for commercial spaceflight. Nat. Med. 28, 611–612 (2022).
Convertino, V. A. Status of cardiovascular issues related to space flight: Implications for future research directions. Respir. Physiol. Neurobiol. 169, S34–S37 (2009).
Hughson, R. L. et al. Cardiovascular regulation during long-duration spaceflights to the International Space Station. J. Appl Physiol.112, 719–727 (2012).
Lathers, C. M. et al. Acute hemodynamic responses to weightlessness in humans. J. Clin. Pharm. 29, 615–627 (1989).
Norsk, P. et al. Fluid shifts, vasodilatation and ambulatory blood pressure reduction during long duration spaceflight. J. Physiol. 593, 573–584 (2015).
Mehta, S. K. et al. Multiple latent viruses reactivate in astronauts during Space Shuttle missions. Brain Behav. Immun. 41, 210–217 (2014).
Capri, M. et al. Long-term human spaceflight and inflammaging: Does it promote aging?. Ageing Res Rev. 87, 101909 (2023).
Lee, A. G. et al. Space Flight-Associated Neuro-ocular Syndrome. JAMA Ophthalmol. 135, 992–994 (2017).
van Oosterhout, W. P. J. et al. Frequency and clinical features of space headache experienced by astronauts during long-haul space flights. Neurology 102, e209224 (2024).
Garrett-Bakelman, F. E. et al. The NASA twins study: a multidimensional analysis of a year-long human spaceflight. Science 364, eaau8650 (2019).
Auñón-Chancellor, S. M. et al. Venous Thrombosis during Spaceflight. N. Engl. J. Med. 382, 89–90 (2020).
Marshall-Goebel, K. et al. Assessment of jugular venous blood flow stasis and thrombosis during spaceflight. JAMA Netw. Open 2, e1915011 (2019).
Kumari, S. et al. A review on saliva-based health diagnostics: biomarker selection and future directions. Biomed. Mater. Devices 2, 121–138 (2023).
Cope, H. et al. Routine omics collection is a golden opportunity for European human research in space and analog environments. Patterns 3, 100550 (2022).
Ruyters, G. & Stang, K. Space medicine 2025 – A vision: Space medicine driving terrestrial medicine for the benefit of people on Earth. REACH 1, 55–62 (2016).
Overbey, E. G. et al. Collection of biospecimens from the inspiration4 mission establishes the standards for the space omics and medical atlas (SOMA). Nat. Commun. 15, 4964 (2024).
Jones, C. W. et al. Molecular and physiological changes in the SpaceX Inspiration4 civilian crew. Nature 632, 1155–1164 (2024).
Brojakowska, A. et al. Retrospective analysis of somatic mutations and clonal hematopoiesis in astronauts. Commun. Biol. 5, 828 (2022).
Chakrabortty, S. K. et al. Exosome based analysis for Space Associated Neuro-Ocular Syndrome and health risks in space exploration. NPJ Microgravity 8, 40 (2022).
Puscas, M. et al. Rare diseases and space health: optimizing synergies from scientific questions to care. npj Microgravity 8, 58 (2022).
Reich, C. et al. OHDSI Standardized Vocabularies-a large-scale centralized reference ontology for international data harmonization. J. Am. Med. Inf. Assoc. 31, 583–590 (2024).
Witze, A. 2022 was a record year for space launches. Nature 613, 426 (2023).
Mason, C. E., et al. A second space age spanning omics, platforms and medicine across orbits. Nature 632, 995–1008 (2024).
Zneimer, S. M. & Hongo, D. Preparing for Clinical Laboratory Improvement Amendments (CLIA) and College of American Pathologists (CAP) Inspections. Curr. Protoc. 1, e324 (2021).
Kalia, S. S. et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 19, 249–255 (2017).
Annaratone, L. et al. Basic principles of biobanking: from biological samples to precision medicine for patients. Virchows Arch. 479, 233–246 (2021).
Williams, S. A. et al. Plasma protein patterns as comprehensive indicators of health. Nat. Med. 25, 1851–1857 (2019).
Ahmad, A., Imran, M. & Ahsan, H. Biomarkers as biomedical bioindicators: approaches and techniques for the detection, analysis, and validation of novel biomarkers of diseases. Pharmaceutics 15, 1630 (2023).
Qiu, S. et al. Small molecule metabolites: discovery of biomarkers and therapeutic targets. Signal Transduct. Target Ther. 8, 132 (2023).
Szilágyi, M. et al. Circulating cell-free nucleic acids: main characteristics and clinical application. Int. J. Mol. Sci. 21, 6827 (2020).
Bezdan, D. et al. Cell-free DNA (cfDNA) and exosome profiling from a year-long human spaceflight reveals circulating biomarkers. iScience 23, 101844 (2020).
Ames, N. J. et al. The human microbiome and understanding the 16S rRNA gene in translational nursing science. Nurs. Res. 66, 184–197 (2017).
René Rohrmanstorfer, S.Z. et al. DNA by Mail: ensure DNA integrity by use of self-drying buccal swabs. Lett. Health Biol. Sci. 2, 1–7 (2017).
Acknowledgements
This study was funded (Grant# INN0010) by the Translational Research Institute for Space Health through NASA Cooperative Agreement NNX16AO69A. The funder played no role in the study design, data collection, analysis and interpretation of data, or the writing of this manuscript. We thank Dr. Jeffrey Rogers for the critical review of this manuscript and Ms. Christie Kovar for advice on the sample de-identification process.
Author information
Authors and Affiliations
Contributions
H.D., E.U.O., and R.A.G. conceptualized the study. S.V.B., S.M.G., D.P.B., H.C., and M.M.M. contributed to biospecimen collections. M.C.G., Y.C., M.M.M., and S.V.B. processed the samples and performed QC. A.K., M.C.G., H.D. prepared the original draft of the manuscript and addressed the edits. V.K., H.M., J.E.P., J.H.W., and D.M. were involved in data generation. M.M. and M.J. contributed to biobank and LIMS development. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Krishnavajhala, A., Gingras, MC., Urquieta, E. et al. The GENESTAR manual for biospecimen collection biobanking and omics data generation from commercial space missions. npj Microgravity 11, 16 (2025). https://doi.org/10.1038/s41526-025-00472-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41526-025-00472-1
