
Abstract
Although many researchers of Parkinson’s disease (PD) have shifted their focus from the central nervous system (CNS) to the peripheral blood, a significant knowledge gap remains between PD severity and the peripheral immune response. In the current study, we aimed to map the peripheral immunity atlas in peripheral blood mononuclear cells (PBMCs) from PD patients and healthy controls using single-cell RNA sequencing (scRNA-seq). Our study employed scRNA-seq analysis to map the peripheral immunity atlas in PD by profiling PBMCs from PD-Early, PD-Late patients and matched controls. By enlarging the blood sample size, we validated the roles of NK cells in numerous immune-related biological processes. We also detected the infiltration of NK cells into the cerebral motor cortex as the disease progressed, using human brain sections, and elucidated the communication between the periphery and CNS and its implications for PD. As a result, cell subpopulation atlases in PBMCs from PD patients and healthy controls along with differentially expressed genes in NK cells were identified by scRNA-seq analysis, representing 6 major immune cell subsets among which NK cells declined in the progression of PD. We further validated NK cell reduction in increasing samples and found that they participated in numerous immune-related biological processes and infiltration into the cerebral motor cortex as the disease proceeded, evidencingd the close communication between the peripheral immune response and CNS. Strikingly, XCL2 positively correlated with PD severity, with good predictive performance of PD and specific expression in subclusters C2 and C5 of NK cells. All these findings delineated the critical role of peripheral immune response mediated by NK cells in the pathogenesis of PD. NK cell-specific XCL2 could be used as a diagnostic marker for treating PD. The indispensable function of NK cells and NK cell-specific molecular biomarkers highlighted the implication of the peripheral immune response in PD progression. Trial registration: ChiCTR, ChiCTR1900023975. Registered 20 June 2019 - Retrospectively registered, https://www.chictr.org.cn/showproj.html?proj=31035.
Similar content being viewed by others


Introduction
Parkinson’s disease (PD) is a common neurodegenerative disease characterized by the accumulation of intracellular protein inclusions, known as Lewy bodies (LBs), which are a pathological marker of sporadic and familial PD. Due to the aging of the population, as many as 12 million people are expected to suffer from PD in the next ten years1. At present, the diagnosis of PD is based on purely clinical observations with limited accuracy and a high misdiagnosis rate2, which has long posed great challenges in clinical medicine. Accumulating evidence shows that biomarkers may potentially be used to diagnose neurodegenerative diseases. Cerebrospinal fluid (CSF) biomarkers are utilized in the diagnosis of PD3, which include biomarkers of CSF Aβ1-42, total tau (T-tau), tau phosphorylated at threonine 181 (P-tau181)4, neurofilament light chain protein (NFL), alpha-synuclein, and heart fatty acid–binding protein (HFABP)5, etc. However, due to the risk and invasiveness of lumbar puncture, the application of these biomarkers in early clinical diagnosis and the monitoring of curative effects is limited.
Many recent neurodegenerative diseases studies have shifted their focus from the central nervous system (CNS) to the peripheral blood6,7. Under physiological conditions, peripheral immune cells are hardly detectable in the CNS due to the existence of a blood-brain barrier (BBB). However, the pathological processes of PD can break down the BBB, leading to increased infiltration of peripheral immune cells into the CNS, which has been identified as one of the major contributing factors for PD8,9. Recently, perturbations of the peripheral immune responses have been unveiled in the disease progression of PD10,11. It has been suggested that T-cell responses associated with α-synuclein pathology may damage key areas of the CNS in PD using a mouse model12. In a recent study, NK cells were shown to be scavengers of α-syn aggregates, and systemic depletion of NK cells in a preclinical PD mouse model exacerbated synuclein pathology13. However, the construction of a peripheral immune response atlas of PD progression is still currently lacking. There is an urgent need to better understand the host immune response atlas during PD progression to decipher prognostic and diagnostic markers and to design appropriate therapeutic interventions for patients with severe disease presentation.
Recently, single-cell RNA sequencing (scRNA-seq) has proven to be an effective strategy to obtain a global view of disease-associated changes at an unprecedented resolution14,15,16,17. ScRNA-seq can enable the identification of disease-specific genetic variants to reveal disease trait associations in specific cell types so that multiple candidate biomarkers can be determined for diagnostic and prognostic use in preclinical and clinical studies17. In this study, differentially abundant cell subpopulation atlases in peripheral blood mononuclear cells (PBMCs) from PD patients and healthy individuals and differentially expressed genes (DEGs) in NK cells were identified by scRNA-seq analysis. The indispensable function of NK cells and an NK cell-specific molecular biomarker highlighted the implication of the peripheral immune response in PD progression. Identifying possible pathogenic cell types and molecular biomarkers underlying PD reveals the feasibility of identifying disease biomarkers in specific cell types rather than in whole blood. It will prompt researchers to develop more effective PD diagnostic means based on scRNA-seq profiling.
Results
ScRNA-seq annotated the cell clusters of the PBMC populations in the healthy controls and PD patients
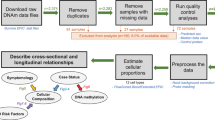
The detailed procedure flow in the present study is shown in Fig. 1A. PBMCs were extracted from healthy controls, PD-Early and PD-Late patients for scRNA-seq at 10× Genomics platform to obtain the cell subsets and differential gene expression information (Fig. 1A, Supplementary Table 1). The number of cells captured in each group was 8269 for sample 16, 7292 for sample 17, 13675 for sample 19, 9005 for sample 20, 4110 for sample 25 and 8647 for sample 26. After quality control, we finally obtained 48523 cells (Fig. 1B, Supplementary Fig. 1A–F, Supplementary Table 2), and the deviation among these groups was assessed, disclosing no significant differences in the average UMI and average gene count within and between groups (Supplementary Fig. 1G–I). We performed clustering analysis to examine cellular heterogeneity in each cell subset from PBMCs. The cell subpopulations were annotated and visualized by UMAP, representing 12 cell clusters of different cell types (Fig. 1C, Supplementary Fig. 2A, Supplementary Table 3). The expression difference of cluster-specific genes was exhibited (Supplementary Fig. 2B), on this basis, 12 clusters were divided into 6 major cell populations based on differential marker gene expression, CD4+ T cells, CD8+ T cells, NK cells, B cells, monocytes and granulocyte-mononuclear progenitor (GMP) cells (Fig. 1D, E, Supplementary Table 4).

A The experimental workflow in the present study. B The number of cells in each sample obtained by 10× scRNA-seq before and after quality control. C UMAP projection of single cells of 12 distinct clusters. D Dot plot shows the average expression of classical marker genes for all 6 cell populations. E UMAP pseudotime representation of 6 major immune cell subsets. CD4_T cells: clusters 0, 6, 8; CD8_T cells: clusters 1, 4, 10; NK cells: cluster 2; monocytes: clusters 3, 7, 9; B cells: cluster 5; GMP cluster 11.
Genetic signatures in major cell subsets and correlation with the risk of PD
In order to elucidate the correlation between peripheral immune cells and PD risk. The critical genes expressed in different types of cells were identified firstly (B cell: CD79A, T cell: CD3D, CD8A and CD4, monocytes: LYZ, NK cell: GNLY, GMP: SOX4) (Fig. 2A). The expression of DEGs in the 6 cell types was unveiled in the heatmap (red indicates upregulation, and green indicates downregulation) (Fig. 2B). These upregulated or downregulated DEGs of the 6 cell groups in the progression of PD (healthy-early-late), and PD pathway-related genes selected from PD-related gene set on GSEA website (https://www.gsea-msigdb.org/gsea/msigdb/human/search.jsp) (Supplementary Table 5), were then imported into a Venn diagram to obtain the intersection. The presence of numerous intersecting DEGs within each cell type suggests a potential correlation between these DEGs and PD risk (Fig. 2C). These findings underscored the close association of peripheral immune cells with PD (Fig. 2D).

A UMAP plot shows the expression of classical marker genes for the 6 cell subsets. B The heatmap shows differential expression of DEGs in different cell types at different stages of PD. C The intersections of upregulated or downregulated DEGs in different cell subsets with genes in the PD pathway. D Cartoon images illustrate potential relationship between peripheral immune cells and PD.
The comparison on changes of 6 cell populations in each sample was displayed in UMAP plots (Fig. 3A, B). Collectively, the quantification data showed a high cell density of T cells (CD8_T cell, CD4_T cell) (clusters 0, 1, 4, 6, 8 and 10) and NK cells (cluster 2) in each sample (Fig. 3C). In the PBMCs of individuals with early and advanced stages of PD, there was a reduction trend in the proportion of NK cells compared to healthy controls, alongside an initial increase followed by a subsequent decrease in T cell proportions in contrast to healthy controls (Fig. 3C, D, Supplementary Table 4).

A UMAP plots show the distribution of major cell subsets in each sample. B The distinct changes of major cell subsets among healthy controls, early and late PD patients. C, D Bar charts display the percentages of major cell subsets in each sample at different stages of PD.
Verification of the varying regularity of NK cells in human peripheral blood and brain during PD progression
Given the scRNA-seq outcomes, we enlarged the sample inclusion by collecting PBMCs from 14 healthy controls, 28 PD-Early patients and 14 PD-Late patients (Supplementary Table 6). T cells, B cells and NK cells in the PBMCs were sorted by flow cytometry. CD45+ (FITC+) cells were selected as R2, from which CD45+CD3+ cells were selected as R3 (T cells). From R2, CD19+ (ECD) and CD3− cells (PC5−) were identified as R4 (B cells), and CD16+ cells (PE), CD56+ cells (RD1) and (PC5−) were selected from R2 as R5 (NK cells) (Fig. 4A, Supplementary Fig. 3A). The results showed that the proportions of NK cells in the PBMCs of PD patients were decreased, with prominent reduction in PD-Late patients, and marked negative correlation was revealed between proportional decline of NK cells and PD severity (Fig. 4A–C). No obvious change was observed in B cell numbers among 3 groups. Despite a significant reduction of T cell numbers in PD-Late patients, the proportion of T cells did not exhibit a substantial correlation with PD severity in our outcomes (Fig. 4A–C). To exclude the interference of sex in cellular proportion, we analyzed the cell proportions of T cells, B cells and NK cells in males and females among healthy controls, PD-Early and PD-Late patients and found no apparent difference in cell proportions between male and female individuals (Supplementary Fig. 3B–E). The decrease in NK cells was further evidenced by the significantly diminished expression of CD16 and CD56 (marker genes of NK cells) in the PBMCs of patients (Fig. 4D, E). These findings highlighted the possible correlation of NK cells with PD progression, which prompted us to consider if NK cells can infiltrate into the brains to influence PD pathology. Thus, further validation on NK cells was performed using human brains sections (Supplementary Table 7). The results first demonstrated visible degeneration and a significant decrease of tyrosine hydroxylase (TH intensity) in the amygdala and motor cortex of PD-Late patients (Fig. 4F, G). Strikingly, we found the existence of NK cells in the amygdala and motor cortex of PD-Late patients, and there is a trend of increasing numbers in the motor cortex (Fig. 4H, I). Furthermore, reduction of TH intensity exhibited significant negative correlation with increase of NK cells amount in the amygdala and motor cortex of PD-Late patients (Fig. 4J). These results indicated that NK cells may infiltrate into the brains and play an indispensable role in the pathology of PD.

A, B Scatter plots and bar graphs of flow cytometry demonstrates the percentages of T cells (CD45+CD3+), B cells (CD45+CD3-CD19+) and NK cells (CD45+CD3-/CD16+CD56+) in the PBMCs of healthy controls (n = 14), PD-Early (n = 26) and PD-Late patients (n = 13–14). C Bar charts show the correlation of the percentage of T cells, B cells and NK cells with HY scores of PD patients. D, E Immunostaining and quantification show the expression of NK cells (CD16+ and CD56+) in human PBMCs at healthy controls (n = 6), early (n = 4–6) and late (n = 5–6) stages. Green, CD56 and CD16 positive cells; blue, DAPI. Scale bar = 50 μm. Data were presented as mean ± s.d. and analyzed by one-way ANOVA followed by Tukey’s post hoc test. F, G Immunostaining of TH in the amygdala and motor cortex of healthy controls (n = 3 individuals) and PD-Late (n = 5 from 2 patients) human brain sections. Scale bar = 50 μm. Green, TH positive cells; blue, DAPI. Data are presented as mean ± s.d. and analyzed using two-tailed pairwise Student’s t test. H, I Immunostaining and quantification of NK cells in the amygdala and motor cortex of healthy controls (n = 6–8 from 3 individuals) and PD-Late (n = 3–5 from 2 patients) human brain sections. Green, CD56 positive cells; blue, DAPI. Data are presented as mean ± s.d. and analyzed using two-tailed pairwise Student’s t test. J Correlation of NK cells amount with TH intensity in the amygdala and motor cortex of PD-Late patients. *p < 0.05, **p < 0.01, ***p < 0.001, ns represents no significance.
NK cells play an important role in the progression of PD based on bioinformatics analysis
The biological functions of NK cells in PD were further explored by bioinformatics analysis. These DEGs in NK cells, which were consecutively upregulated in the progression of PD, were sorted into a heatmap (Fig. 5A). The upregulated DEGs at different stages were intersected to screen pivotal genes, and a total of 200 pivotal genes were overlapped (Fig. 5B). GO analysis yielded the top ten enriched BP, MF, and CC terms involved by those overlapped genes (Fig. 5C), revealing that most of these terms were immune-related. Among those upregulated DEGs, more crucial ones were further identified by the volcano map and heatmap (Fig. 5D, E). GO analysis revealed that these crucial upregulated DEGs were associated with immune regulation and cytokine responses (Fig. 5F), suggesting that NK cells may play an important role in the immune regulation of PD. In addition, a similar analysis was performed for downregulated DEGs in NK cells (Fig. 5G), intersecting 202 consecutively downregulated genes during the disease process (Fig. 5H). GO analysis of these DEGs revealed that they were mainly associated with DNA transcription and signaling (Fig. 5I). KEGG analysis identified the most striking MAPK signaling pathway involving these downregulated DEGs (Fig. 5J). The hub genes identified by PPI analysis (C5AR1, GNG7, PPBP, CXCR3, PF4, UBR1, FBXO11, TRAF7, KCTD6) could provide a basis for subsequent studies on related functions as well as screening pathways (Supplementary Fig. 4A). The disease-related risk scoring also showed a correlation of NK cells with PD risk (Supplementary Fig. 4B). The PD risk genes were further intersected with the upregulated and downregulated DEGs in NK cells, and the overlapped upregulated DEGs were also involved in immune regulation processes (Supplementary Fig. 4C–E). Comparatively, we also selected DEGs continuously downregulated in CD8+ T cells from PBMCs of PD patients, and 89 hub genes were obtained after the intersection (Supplementary Fig. 4F). PPI analysis, GO and KEGG were performed on these 89 hub genes, which revealed a close relationship with neurogenerative diseases (Supplementary Fig. 4H, I), consistent with previously published studies18,19. Therefore, our subsequent studies mainly focused on NK cells to explore biological markers in the progression of PD.

A Heatmap shows the expression of the upregulated DEGs in NK cells at healthy control, PD-Early and PD-Late stages. B Intersection of three sets of fold change values. FC1 (Fold change 1), PD-Early vs. healthy control; FC2 (Fold change 2), PD-Late vs. PD-Early; FC3 (Fold change 3) PD-Late vs. healthy control. C GO analysis of the intersected hub genes. D Volcano plot displays differential expression of the intersected hub genes in NK cells. E Heatmap shows the expression changes of the crucially upregulated DEGs obtained by volcano plot at healthy control, PD-Early and PD-Late stages. F GO analysis of the crucially upregulated DEGs in NK cells obtained by volcano plot. G Heatmap displays the expression variation of downregulated DEGs at healthy control, PD-Early and PD-Late stages. H Intersection of three sets of fold change values. FC1 (Fold change 1), PD-Early vs healthy control; FC2 (Fold change 2), PD-Late vs PD-Early; FC3 (Fold change 3), PD-Late vs healthy control. I GO analysis of the intersected hub genes. J KEGG enrichment analysis of the intersected hub genes.
Identification and verification of NK cell-specific PD-associated crucial genes
Given the immune regulation of NK cells in PD, we screened the most crucial DEGs related to the immune pathway and cytokine response out of the aforementioned upregulated DEGs so as to determine crucial genes in NK cells that might play a vital role in the progression of PD. Excluding the genes associated with hemoglobin (HBB), cytoskeleton (ACTB) and lymphotoxin (PSME2, LTB) in the blood, we verified the remaining 9 genes related to signal transduction and immune regulation by RT‒qPCR (Fig. 6A, B). Of the 9 DEGs, only JUN and XCL2 were significantly upregulated in PD (Fig. 6B). In clinical efficacy evaluation, the area under the ROC curve (AUC) (95% confidence interval) of JUN for the prediction of PD 2.0, PD 3.5 and PD were 0.7222 (p = 0.0551), 0.6857 (p = 0.2046) and 0.6080 (p = 0.3242), while the AUC of XCL2 for the prediction of PD were 0.7556 (p = 0.0274), 0.9286 (p = 0.0034) and 0.8040 (p = 0.0055) (Fig. 6C). This implied the predictive performance of XCL2 is superior to JUN. In addition, we further compared the relative expression of 9 DEGs in males and females and found no apparent difference in gene expression between them, except in MT2A (Supplementary Fig. 5A). Moreover, the predictive performance of JUN (AUC, 0.5667) and XCL2 (AUC, 0.5635) did not exhibit any significant difference between male and female patients (Supplementary Fig. 5B). Thus, sex difference was not considered in the study design. The expression of JUN and XCL2 in different cell populations was analyzed, which revealed that JUN existed in B cells, NK cells and monocytes, but that XCL2 was specifically expressed in T cells and NK cells, particularly with increased expression in NK cells as the disease progressed (Fig. 6D, E). Additional correlation analysis of JUN and XCL2 in NK cells showed that XCL2 significantly correlated with the HY scores of PD patients (Fig. 6F, G). These results suggested that the specific differential expression of XCL2 in NK cells may be a key marker in PD patients.

A Screening the DEGs in NK cells. B Relative expression of the selected NK cell-specific upregulated DEGs. All healthy controls (n = 13), PD (n = 36, CCL3; 31, NFKBIA; 35, JUN; 29, IER2; 25, IER2; 38, LY6E; 38, MT2A; 35, XCL2; 35, FOS; 35, IFITM3). C AUC values of JUN and XCL2 for prediction of PD individuals at different stages. D The expression of JUN in major cell subsets of PBMCs at healthy control, early and late PD stages. E The expression of XCL2 in major cell subsets of PBMCs at healthy control, early and late PD stages. F Correlation of JUN levels with HY scores of PD patients. G Correlation of XCL2 levels with HY scores of PD patients.
Specific biomarker XCL2 in the subgroups of NK cells in the progression of PD
The subsets of NK cells were further analyzed and 8 NK subgroups (C0-C7) were ultimately identified, with the average number of cells in these 8 subgroups reaching 995 (163-2684) (Fig. 7A). By analyzing the specific marker genes of these 8 subgroups, we found that C2 and C5 subgroups are specifically overexpressing XCL2 (Fig. 7B, C). After standardizing the cell count, it was found that compared to healthy controls, the C2 subgroup increased in the early stages of PD and maintained a higher proportion during disease progression; C5 subgroup showed a mild increase in the early stage of PD and an increase in the late stage, indicating that XCL2 positive NK cell subgroups (C2 and C5) may be associated with the occurrence and progression of PD (Fig. 7D). Based on cell density visualization, XCL2 was highly enriched in the C2 and C5 subgroups, and differential gene analysis showed that XCL2 was more expressed in the C2 and C5 subgroups of PD patients compared to healthy controls (Fig. 7E, F). Further, GO enrichment and enrichment network analysis were performed on the specific marker genes of C2 and C5 subgroups. We found that genes with specific high expression in C2 are mainly related to biological functions such as positive regulation of T cell activation, interferon-beta production, interferon-gamma production, and chemotherapy mediated signaling pathway. Genes with specific high expression in C5 are mainly related to biological functions such as gamma delta T cell activation and interleukin-12 production (Fig. 7G). XCL2 in disease-related NK subpopulations is involved in these biological functions, further indicating its important role in the occurrence and development of PD.

A UMAP shows the divided 8 clusters of NK cells. B The expression of gene markers used to identify the 8 clusters of NK cells. C UMAP plots display the expression distribution of classic markers of NK cells and specific markers of NK subpopulations. D The proportions of 8 NK subclusters at healthy control, early and late stages. E UMAP plots shows the enrichment of XCL2 expression in different NK subclusters. F Dot plot illustrates XCL2 expression in healthy control, early- and late-stage PD patients between C2 and C5 subclusters. G GO enrichment analysis and enrichment network analysis demonstrate the biological pathways involved by specific genes with high expression in C2 and C5 subgroups.
Discussion
In this study, we mapped the peripheral immune atlas in PD by scRNA-seq and defined specific PD diagnostic markers, including 6 major immune cell subsets, T cells (CD4+, CD8+), B cells, NK cells, monocytes and GMP, and displayed the variation rules of cell subsets and different gene expression patterns in the progression of PD. We further enlarged the sample size, and through clinical validation, we found that among these cell subsets, NK cells were involved in numerous immune-related biological processes and showed a significant decrease in blood samples with the progression of PD. As the disease progresses, NK cells might infiltrate into the motor cortex of the brain, proving close communication between the peripheral immune response and the CNS. Moreover, XCL2 was explicitly expressed in sub-cluster C2 and C5 of NK cells, which might become a specific diagnostic marker for PD.
The peripheral immune system plays an important role in the development of PD. Numerous studies have shown that immune cells, particularly lymphocytes, are involved in the pathogenesis of PD20,21,22,23. In the present study, we constructed a peripheral immunity atlas of PD and revealed the variation in cell subsets, which showed that the number of NK cells declined in peripheral blood as PD proceeded and was involved in immune-related biological processes. As known in the innate immune response in PD, in addition to microglia, blood monocytes, neutrophils, and NK cells are involved in this process, as suggested by transcriptomic and cellular analyses of blood from people with PD24,25. Many existing studies have already highlighted the relevance of peripheral monocytes in PD risks26,27. People with PD had increased monocyte numbers in the CSF, and blood monocytes antigen-presenting (HLA-DR) and phagocytic capacity (CD163) increased with time, becoming significant at later PD stages26. Consistently, our scRNA-seq outcomes exhibited a similar higher proportion of monocytes at a late stage of PD than that at the early stage, providing valid evidence at the single cell level for monocytes investigations in PD. However, fewer have reported the relevance of NK cells in PD, whose infiltration is also involved in the inflammatory process and substantia nigra neurodegeneration13. In this study, in addition to the proportional reduction of NK cells at early and late stages revealed by scRNA-seq data, disease-related risk scoring also revealed a correlation of NK cells with PD, and the PD risk genes in NK cells were also related to immune regulation. These results implied that abnormal changes in NK cells lead to immune dysregulation in the peripheral blood of PD patients, thus aggravating the severity of the disease.
Our results about NK cells in PD patients have also revealed enrichment in mitochondria related processes. In previous reports, dopaminergic neurons in PD patients typically exhibit mitochondrial dysfunction, leading to insufficient energy metabolism and increased oxidative stress, exacerbating neuronal damage28,29. Mitochondrial-produced ATP is a crucial energy source for maintaining the function of both neurons and immune cells. During inflammatory states, the activation and proliferation of immune cells require significant energy, which mitochondria adjust through metabolic adaptations to meet this demand. Simultaneously, alterations in energy supply can impact neuronal function and survival, thereby influencing neuroimmune inflammatory responses30. Consistently, our outcomes highlighted the critical role of NK cells in the progression of PD.
Close communication exists between the peripheral immune response and the CNS10. It has been widely observed that NK cells infiltrate the brain in neurodegenerative deficits such as Alzheimer’s disease (AD) and PD13,31 and regulate the expression of immunity-related genes in peripheral NK cells, which can promote the NK cell infiltration in the brain and neuroinflammatory changes in AD31. In parallel, studies in PD animal models, mainly α-syn driven, suggest that infiltrating NK cells are involved in the inflammatory process and substantia nigra neurodegeneration since removing these cellular compartments affects neuronal survival13. We speculated that during PD progression, the integrity of the BBB is compromised, thus causing activated resident innate immune cells within the brain, including microglia and astrocytes, to recruit peripheral innate immune cells. These results strengthen the implication of NK cells in the peripheral immune system in deteriorating the immune environment in the CNS. In addition, it is well known that PD patients have obvious characteristic motor symptoms, such as bradykinesia and myotonia, which are closely related to the motor cortex of the brain. The present study using human brain samples at an advanced stage suggested that peripheral NK cells may infiltrate into the brain motor cortex and amygdala, potentially participating in PD pathogenesis. Recent investigations reported obvious motor cortical thinning and amygdala volume loss in severe PD, and these morphometric changes are associated with longer disease duration and increasing motor symptom severity32, which could have implications for the future development of PD impairments. Further validation and in-depth mechanistic exploration of NK cells in a larger cohort of clinical subjects are essential in advancing PD research, necessitating the increase of clinical samples and the execution of comprehensive assays.
Moreover, we tried to explore the cell-specific immune-related biomarkers specifically expressed in NK cells during PD progression. Our data showed that the prominently differential expression of XCL2 in NK cells performed well in predicting different stages of PD and had a significant positive correlation with the severity of PD patients. Based on this, we further analyzed the subgroups of NK cells, and were surprised to find that, with the increase in PD severity, XCL2 showed prominently augmented levels in subclusters C2 and C5 of the NK cells. This observation suggested that NK cell subtypes C2 and C5 may be the predominant cell types infiltrating the brain in PD, potentially involved in immune responses within the brain. Further research could explore the exact functions and mechanisms of these specific NK cell subtypes in PD pathogenesis. As previously reported, XCL2 transcripts are closely correlated with the gene signatures of NK cells and are associated with overall patient survival in cancers33. Few current studies have illustrated the characterization of NK cell-specific XCL2 expression in PD development. It has been reported that human NK cells can produce the chemokines CCL5, XCL1, and XCL2 but are a rare source of the latter two in blood under steady-state conditions33, which can explain the abnormal upregulation of XCL2 in peripheral NK cells that was reduced as PD proceeded. These revealed critical features of peripheral immune responses in the pathogenesis and progression of PD and identified NK cell populations with specialized gene expression programs.
In conclusion, we mapped the peripheral immunity atlas in PD by scRNA-seq and revealed variation laws of cell subsets and different gene expressions in the progression of PD. Further, the infiltration of NK cells in the brain motor cortex as the disease progressed demonstrates the close communication between the peripheral immune response and the CNS. Strikingly, NK cell-specific XCL2, which shows a positive correlation with PD severity and demonstrates strong predictive capabilities for PD, promises to offer valuable insights and potential therapeutic targets for future PD treatment strategies.
Materials and methods
Participants
According to the maximum available clinical data, we first included 4 PD patients at different stages who were diagnosed in the West China Hospital of Sichuan University (Sichuan, China) from September 2017 to March 2018 with the following inclusion criteria: a clinical diagnosis of PD-Early (Hoehn-Yahr (HY) stages I-II)34 or PD-Late (HY stages III-IV), and Unified Parkinson’s Disease Rating Scale Part-III (UPDRS-III) motor severity scores32 (PD-Early, 24.96 ± 4.88; PD-Late, 44.15 ± 4.16), aged 60–90 years, and stable administration of medication. None of the patients had significant somatic disorders, such as tumor, hypertension, diabetes, autoimmune disorders and chronic diseases, as well as psychiatric co-morbidities, including mild cognitive impairment (MCI) and dementia. The family history does not suggest any hereditary factors. Two age-matched healthy controls were also recruited, and they did not suffer from neurodegenerative, neurogenetic and movement disorders. Detailed information on the involved individuals is available in Supplementary Table 1. All the participants were informed of the study significance and signed written informed consent forms. PBMCs from these 6 participants was collected for scRNA-seq analysis to obtain the cell subsets and differential gene information. Referring to these inclusion criteria and the MedSci Sample Size calculation method, we subsequently continued to expand samples by collecting venous blood from 42 PD patients (28 early, 14 late) and from 14 healthy subjects from September 2017 to January 2021 for fluorescence-activated cell sorting (FACS), correlation analysis with the patients’ HY scores and further PCR detection to verify the changes in differential cell populations and the expression of candidate molecules in the PBMCs of PD patients. In addition, postmortem human brain samples from histologically confirmed cases of PD and age-matched healthy individuals without dementia were also used (Supplementary Table 7). All research procedures involving human subjects were clinically registered and approved by the Clinical Trial and Biomedical Ethics Committee of the West China Hospital of Sichuan University (approval number 20150236; Chinese Clinical Trial Registry number ChiCTR1900023975). Additionally, this study was conducted under the guidelines of the Declaration of Helsinki.
Isolation and culture of PBMCs
Patient blood is collected at the same time in the morning. After being stored at 4 °C, PBMCs were transported to the laboratory and separated and extracted within 4 h. PBMCs were isolated from blood samples of healthy controls and PD patients (PD-Early and PD-Late) and purified by Ficoll gradient separation (Ficoll–PaqueTM Plus, GE Healthcare, Sweden). Briefly, a 3 mL Ficoll-Parker gradient was aspirated into two 15 mL centrifuge tubes. Blood samples were diluted at a ratio of 1:1 with phosphate-buffered saline (PBS) and then layered over the Ficoll-Parker gradient, subsequently followed by centrifugation at 400 × g for 30 min. The cells were collected carefully from the interface layer between the plasma and Ficoll-Paqeu media (8 mL/tube) and washed twice in PBS (at 100 × g for 10 min). Upon isolation of PBMCs from collected blood samples, we immediately cryopreserved the cells using DMEM high-glucose medium supplemented with 20% serum and 15% DMSO freezing solution. The cells were counted using a trypan blue haemocytometer and were frozen in a liquid nitrogen tank at a density of 3 ×106 cells/tube.
10× Genomics single-cell packaging and library preparation
As previously described35, 10× Genomics Chromium Next GEM Single Cell 3ʹ Reagent Kits v3.1 (10× Genomics, 1000268) were used to generate a single-cell library according to the manufacturer’s protocol (Oebiotech, Shanghai). PBMCs were suspended in 0.04% BSA-PBS and then were counted with a Countess® II Automatic Cell Counter. We adjusted the cells to the desired concentration of 1 ×106/mL. Approximately 13000 cells were added to each channel, and nanoliter-scale gel bead-in-emulsions (GEMs) containing barcode information were generated and then reverse transcribed in the C1000 Touch Thermal Cycler (Bio-Rad). Then, a Biorruptersonicator was used to fragment the amplified cDNA into approximately 200–300 bp, and traditional second-generation sequencing methods were employed, such as sequencing linker P5 and sequencing primer R1. Finally, the DNA library was amplified by PCR.
Single-cell RNA sequencing
Qubit was applied to quantitatively analyze the library, and the qualified library was placed in cBot for bridge amplification. All the libraries constructed in this study were sequenced on a Illumina Nova 6000 PE150 platform. With 150 bp paired ends, each library was sequenced for a whole lane. Every cycle was supplemented with 4 fluorophore-labeled A, T, G, and C dNTPs. According to the pairing of AT and GC, DNA polymerase was used to combine the corresponding dNTPs with the template DNA strand and to wash other unbound dNTPs. When it is removed, the bound position releases a fluorescence signal, which can be captured and converted by the computer; thus, the base information of the binding position is obtained. The Cell Ranger software pipeline (version 2.2.0) provided by 10× Genomics was used to demultiplex cellular barcodes, to map reads to the genome and transcriptome using the Spliced Transcripts Alignment to a Reference (STAR) aligner, and to downsample reads as required to generate normalized aggregate data across samples, producing a matrix of gene counts versus cells. We processed the unique molecular identifier (UMI) count matrix using the R package Seurat36 (version 2.3.4).
Quality control
To remove low-quality cells and likely multiplet captures, which is a major concern in microdroplet-based experiments, we filtered out cells with UMI/gene numbers that were more than 4 standard deviations away from the mean value, assuming that the UMI/gene numbers of the cells followed a Gaussian distribution. Following visual inspection of the distribution of cells by the fraction of mitochondrial genes expressed, we further discarded low-quality cells in which >10% of the counts belonged to mitochondrial genes. In accordance with quality control (QC) criteria, 48523 single cells (2475 cells filtered out from a total of 50998 cells) remained and were included in downstream analyses (Supplementary Table 2). Library size normalization was performed in Seurat on the filtered matrix to obtain the normalized count.
Data processing
The raw image data files obtained by single-cell RNA sequencing were converted into the original sequences by base-calling analysis. The resulting data were called raw data or raw reads. These results were stored in the FASTQ (.fq) file format, which contains the sequence information (reads) and its corresponding sequencing quality information. The top variable genes across single cells were identified using the method described by Macosko et al. 37. Briefly, the average expression and dispersion were calculated for each gene, and the genes were subsequently divided into 12 bins based on expression. Principal component analysis (PCA) was performed to reduce the dimensionality of the log-transformed gene-barcode matrices of the top variable genes. Cells were clustered based on a gene-based clustering approach and visualized in UMAP using “RunUMAP” function. The cell type signature genes are listed in Table 1, and the number of each cell subset is shown in Supplementary Table 3. Subpopulations of T and NK cells were analyzed in the same way. A likelihood ratio test that simultaneously tests for changes in mean expression and in the percentage of expressed cells was used to identify DEGs between clusters. The R package SingleR, a novel computational method for unbiased cell type recognition of single-cell RNA-seq, was used to infer the cell origin of each single cell independently and identify cell types by referring to the transcriptomic datasets “Blueprint Epigenomics”38 and “Encode”39. DEGs were identified using the Find Markers function (test. use =MAST) in Seurat, and Pearson correlations between samples were calculated in R (cor (data, method = “pearson”)). The criteria of p value < 0.05 and |log2(fold change)|> 0.26 were set as the thresholds for significantly differential expression. Heatmaps, volcano plots, and violin plots were generated using R. The enriched pathways associated with the DEGs were analyzed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (https://www.kegg.jp/). Venn diagrams and circular heatmaps were generated with TBtools (https://doi.org/10.1016/j.molp.2020.06.009).
Expression distance analysis
The expression differences between matching subpopulations were determined by the “estimateExpressionShiftMagnitudes()” function from Cacoa. In short, the expression distance between samples were determined as a normalized weighted sum of the related distances of all cell subpopulations contained in both samples, with a weight equal to the subpopulation ratio (measured as the smallest proportion represented by a given cell subpopulation in the two samples being compared). The expression distance between samples is projected into a two-dimensional (2D) space using a multidimensional scale.
Gene set score analysis
Gene sets related to “Parkinson’s disease” were obtained from the DisGeNET database (https://www.disgenet.org/home/). Gene set scores were acquired by analyzing the transcriptome of each input cell against the aforementioned gene sets using the Seurat function “AddModuleScore”. Changes in the scores among different cell groups were analyzed using the ggpubr R package via the Wilcoxon test.
Fluorescence-activated cell sorting (FACS)
PBMCs from 42 PD patients and 14 healthy control subjects were isolated by FACS to further validate the results of scRNA-seq. Detailed information on the involved individuals is available in Supplementary Table 6. Briefly, all PBMCs were washed, counted and suspended in ice-cold PBS, and cell-surface antigen staining was subsequently performed. Monoclonal antibodies against CD3, CD45, CD56, CD19 (CD45/CD56/CD19/CD3 detection kit, Beckman Coulter, USA) and CD16 (CD16-PE, Beckman Coulter, USA) were used for immunofluorescence staining. The stained cells (T cells (%): CD3+/ all cells excluding dead cells, B cells (%): CD19+CD3− / all cells excluding dead cells, NK cells (%): CD16+CD56+CD3-/ all cells excluding dead cells) were analyzed and sorted by BD Influx (BD, USA), and data were analyzed using FlowJo (version 10.0). Detailed gating strategies were provided as follows: 1) Initial gating to exclude dead cells: we utilized forward scatter height (FSC-A) and side scatter height (SSC-A) parameters to gate out dead cells, resulting in Region 1 (R1); 2) Identification of CD45+ cells: within R1, we selected cells positive for CD45 (FITC+), resulting in Region 2 (R2); 3) Isolation of CD45+CD3+ cells (T cells): from R2, we gated out CD45+CD3+ cells (PC5+), resulting in Region 3 (R3), representing T cells; 4) Isolation of CD19+ cells (B cells): within R2, we gated out cells positive for CD19 (ECD) and cells negative for CD3 cells (PC5-), resulting in Region 4 (R4), representing B cells; 5) Isolation of CD56+CD16+ cells (NK cells): within R2, we selected cells positive for CD56+CD16+ (RD1, PE) and cells negative for CD3 cells (PC5-), resulting in Region 5 (R5), representing NK cells.
Immunofluorescence staining
Human PBMC smears and brain sections were prepared for immunofluorescence staining. The brain slice information is available in Supplementary Table 7. Brain sections firstly underwent antigen retrieval with sodium citrate, and then were washed 3 times with 0.01 M PBS for 5 min each. Afterwards, brain sections and cell smears were then incubated with 5% goat serum and 0.3% Triton X-100 for 2 hours at room temperature. Afterwards, primary antibodies (CD16, CD56, rabbit, Bioss, 1:50; TH, rabbit, Abcam, 1:250) diluted in 2% goat serum were added in the indicated subjects and incubated for 18 h at 4 °C. Sections of negative controls were treated with 2% goat serum. After 3 washes with PBS, sections were incubated with secondary antibody (DyLight 488, goat anti-rabbit IgG, Abbkine, 1:400) for 3 h at room temperature, rinsed 3 times with PBS and stained with DAPI. Finally, the sections were sealed with Antifade Mounting Medium (P0126, Beyotime). Fluorescence changes in the brain tissue were observed under a light microscope. ImageJ software was employed to quantify the cell number of five randomly selected fields (1.33 mm2) separately in the green fluorescence channel (positive cells) and the blue fluorescence channel (DAPI). Subsequently, we calculated the positive rate as: (the number of positive cells/the number of DAPI-stained nuclei) ×100. TH intensity of was measured by ImageJ software, and the positive area of TH was normalized to the DAPI-positive area (3 slices (1.33 mm2) in each group).
Real-time quantitative PCR (RT-qPCR)
We further verified the differences in gene expression of the 9 crucially upregulated DEGs expressed in NK cells using RT-qPCR. Briefly, total RNA from flow-sorted NK cells was extracted using RNAiso plus (TaKaRa, Japan) following the manufacturer’s protocol and reverse transcribed into complementary DNA (cDNA) by the Bestar™ qPCR RT Kit (DBI, Germany). RT-qPCR was performed using a CFX96™ Real-Time System (Bio-Rad, USA). The expression level of the target gene was normalized to the GAPDH level in each sample. Primer sequences are listed in Table 2.
Classification of NK subtypes in PBMCs
We grouped cell clusters with strong signals of GNLY, GZMB and NKG7 expression into NK cells. NK cells were isolated for subpopulation identification. We normalized the raw UMI counts using the “NormalizeData” function in the Seurat with the scaling factor equal to 10,000. Then, the top 3000 highly variable genes were obtained by the Seurat function “FindVariableFeatures” with the default variance stabilizing process. We further integrated nuclei on the basis of the summarized anchor features via the function “IntegrateData” and embedded ensuing nuclei in the PCA dimensions followed by UMAP visualization. Based on ElbowPlot and JackStraw, we choose the appropriate dimension reduction (dims = 20). After multiple “FindClusters” analysis, we chose the most appropriate resolution (resolution = 0.2). NK clusters were first classified to major groups based on the expression of two canonical function markers FCGR3A/CD16 and NCAM1/CD56). “FindMarkers()” function implemented in Seurat was used to identify cluster specific marker genes with the options “logfc.thresh old = 0.25, min.pct = 0.25”. P-value was corrected using the Bonferroni method, and 0.05 was set as a threshold to define significance. Based on the difference of marker gene expression, it can be divided into different subtypes.
Statistical analysis
The sequencing data were analyzed using R software (Version 2.3.4) and Cell Ranger. All data are presented as the primary data. Statistical analysis was performed using SPSS Statistics 25.0 (IBM, Chicago, IL, USA). Independent-sample t tests were used to analyze between-group differences, and comparisons among multiple groups were tested using one-way analysis of variance (ANOVA) with a post hoc test. If the variance was homogeneous, the least significant difference (LSD) result in the multiple comparisons was selected; if not, Dunnett’s T3 test was selected. The Kruskal–Wallis check was used for multiple cluster comparisons that failed to change to traditional homogeneity and homogeneity of variance. The clinical efficacy evaluation data of JUN and XCL2 were analyzed by Kruskal–Wallis test. The RT-qPCR results were used for receiver operating characteristic (ROC) curve analysis. Spearman correlation analysis was utilized for correlation of XCL2 and JUN with PD severity, as they did not meet the normality assumption. For correlation of NK, T, and B cell data with PD severity, along with the correlation between NK cells amount and TH intensity, which adhered to normal distribution, Pearson correlation analysis was applied. Independent sample t test was used to exclude the interference of gender. A Venn diagram was drawn using the TBtools (a Toolkit for Biologists integrating various biological data-handling tools) program (https://doi.org/10.1016/j.molp.2020.06.009)40. R software, the cluster Profile package41 was used for KEGG and GO analysis, and the heatmaply package42 was used for the heatmap. The coexpression of hub genes was analyzed on STRING online (https://string-db.org/) and displayed with Cytoscape software (https://cytoscape.org/). All images and every element within these images were created entirely using Adobe Illustrator, GraphPad Prism, R software. No third-party elements were used in the creation of these figures.
Data availability
The scRNA-seq data reported in this article have been deposited in the Gene Expression Omnibus (GEO) of National Center for Biotechnology Information (NCBI), under accession number GSE223138 and are accessible at with token number (kvijgekgjzcpzid).
References
Wijeratne, T., Grisold, W., Trenkwalder, C. & Carroll, W. World Brain Day 2020: move together to end Parkinson’s disease. Lancet Neurol. 19, 643 (2020).
Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376 (2008).
Hall, S. et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch. Neurol. 69, 1445–1452 (2012).
Kang, J. H. et al. Association of cerebrospinal fluid β-amyloid 1-42, T-tau, P-tau181, and α-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol. 70, 1277–1287 (2013).
Bäckström, D. C. et al. Cerebrospinal Fluid Patterns and the Risk of Future Dementia in Early, Incident Parkinson Disease. JAMA Neurol. 72, 1175–1182 (2015).
Atashrazm, F. et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci. Rep. 8, 15446 (2018).
Papagiannakis, N. et al. Lysosomal alterations in peripheral blood mononuclear cells of Parkinson’s disease patients. Mov. Disord. 30, 1830–1834 (2015).
Pan, Y. & Nicolazzo, J. A. Impact of aging, Alzheimer’s disease and Parkinson’s disease on the blood-brain barrier transport of therapeutics. Adv. Drug Deliv. Rev. 135, 62–74 (2018).
Sweeney, M. D., Sagare, A. P. & Zlokovic, B. V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150 (2018).
Vida, C. et al. Lymphoproliferation Impairment and Oxidative Stress in Blood Cells from Early Parkinson’s Disease Patients. Int. J. Mol. Sci. 20, https://doi.org/10.3390/ijms20030771 (2019).
Kip, E. & Parr-Brownlie, L. C. Reducing neuroinflammation via therapeutic compounds and lifestyle to prevent or delay progression of Parkinson’s disease. Ageing Res. Rev. 78, 101618 (2022).
Williams, G. P. et al. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain J. Neurol. 144, 2047–2059 (2021).
Earls, R. H. et al. NK cells clear α-synuclein and the depletion of NK cells exacerbates synuclein pathology in a mouse model of α-synucleinopathy. Proc. Natl Acad. Sci. USA 117, 1762–1771 (2020).
Zeisel, A. et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015).
Grün, D. et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 525, 251–255 (2015).
Treutlein, B. et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375 (2014).
Shalek, A. K. et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 510, 363–369 (2014).
Kimura, K. et al. Resident Memory-like CD8(+) T Cells Are Involved in Chronic Inflammatory and Neurodegenerative Diseases in the CNS. Neurol. Neuroimmunol. Neuroinflamm. 11, e200172 (2024).
Hu, D. Alzheimer’s defense: brain CD8(+) T cells in action. Trends Immunol. 44, 936–937 (2023).
Miki, Y. et al. Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol. Aging 63, 33–43 (2018).
Mutez, E. et al. Involvement of the immune system, endocytosis and EIF2 signaling in both genetically determined and sporadic forms of Parkinson’s disease. Neurobiol. Dis. 63, 165–170 (2014).
Sala, G. et al. Reduced expression of the chaperone-mediated autophagy carrier hsc70 protein in lymphomonocytes of patients with Parkinson’s disease. Brain Res. 1546, 46–52 (2014).
Fernández, B. et al. Centrosomal cohesion deficits as cellular biomarker in lymphoblastoid cell lines from LRRK2 Parkinson’s disease patients. Biochemical J. 476, 2797–2813 (2019).
Craig, D. W. et al. RNA sequencing of whole blood reveals early alterations in immune cells and gene expression in Parkinson’s disease. Nat. Aging 1, 734–747 (2021).
Jensen, M. P. et al. Lower Lymphocyte Count is Associated With Increased Risk of Parkinson’s Disease. Ann. Neurol. 89, 803–812 (2021).
Pillny, C., Nitsch, L., Proske-Schmitz, S., Sharma, A. & Wüllner, U. Abnormal subpopulations of monocytes in the cerebrospinal fluid of patients with Parkinson’s disease. Parkinsonism Relat. Disord. 84, 144–145 (2021).
Li, Y. I., Wong, G., Humphrey, J. & Raj, T. Prioritizing Parkinson’s disease genes using population-scale transcriptomic data. Nat. Commun. 10, 994 (2019).
Tong, B. et al. Targeting dysregulated lipid metabolism for the treatment of Alzheimer’s disease and Parkinson’s disease: Current advancements and future prospects. Neurobiol. Dis. 196, 106505 (2024).
Buoso, C. et al. Dopamine‑iron homeostasis interaction rescues mitochondrial fitness in Parkinson’s disease. Neurobiol. Dis. 196, 106506 (2024).
Venegas, C. et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 552, 355–361 (2017).
Lu, Y., Li, K., Hu, Y. & Wang, X. Expression of Immune Related Genes and Possible Regulatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 12, 768966 (2021).
Wilson, H., Niccolini, F., Pellicano, C. & Politis, M. Cortical thinning across Parkinson’s disease stages and clinical correlates. J. Neurol. Sci. 398, 31–38 (2019).
Böttcher, J. P. et al. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 172, 1022–1037.e14 (2018).
Hoehn, M. M. & Yahr, M. D. Parkinsonism: onset, progression and mortality. Neurology 17, 427–442 (1967).
Xiong, L. L. et al. Single-cell RNA sequencing reveals B cell-related molecular biomarkers for Alzheimer’s disease. Exp. Mol. Med. 53, 1888–1901 (2021).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Macosko, E. Z. et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214 (2015).
Martens, J. H. & Stunnenberg, H. G. BLUEPRINT: mapping human blood cell epigenomes. Haematologica 98, 1487–1489 (2013).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202 (2020).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287 (2012).
Galili, T., O’Callaghan, A., Sidi, J. & Sievert, C. heatmaply: an R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 34, 1600–1602 (2018).
Acknowledgements
This study was supported by Wang Wenyuan Expert Workstation Project of Yunnan Provincial Science and Technology Talent and Platform Program (202205AF150013), Guizhou Provincial Higher Education Science and Technological Innovation Team ([2023]072) and the Program of Science & Technology Department of Sichuan Province (2020YFS0043). It was also supported by Guizhou Province Distinguished Young Scientific and Technological Talent Program (YQK[2023]040) and Zunyi Medical University 12345 Future Talent Training Program-Technology Elite (ZYSE-2021-03). We give great gratitude to OE Biotech Limited, Huai-Yu Su and Ning-Yi Li for their technique supports.
Author information
Authors and Affiliations
Contributions
L.-L. Xiong., Y.-Y.W. and T.-H.W. conceived the overall study and designed experiments. L.-L.Xue., R.-L.D., and R.-Z.N. performed the data analysis, experiments and figure making. J.H., C.-Y.Y., X.-Y.H., J.L., and X.-Y.H. collected the patient information and prepared the samples. L.-R.H. and X.-X.C. contributed to morphological tests and data quantification. L.-L.Xiong. and L.C. wrote and revised the manuscript. All authors have read and approved the final version of manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, LL., Du, RL., Niu, RZ. et al. Single-cell RNA sequencing reveals peripheral immunological features in Parkinson’s Disease. npj Parkinsons Dis. 10, 185 (2024). https://doi.org/10.1038/s41531-024-00790-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-024-00790-3
