
Abstract
Recent advancements in functional genomics have provided an unprecedented ability to measure diverse molecular modalities, but predicting causal regulatory relationships from observational data remains challenging. Here, we leverage pooled genetic screens and single-cell sequencing (Perturb-seq) to systematically identify the targets of signalling regulators in diverse biological contexts. We demonstrate how Perturb-seq is compatible with recent and commercially available advances in combinatorial indexing and next-generation sequencing, and perform more than 1,500 perturbations split across six cell lines and five biological signalling contexts. We introduce an improved computational framework (Mixscale) to address cellular variation in perturbation efficiency, alongside optimized statistical methods to learn differentially expressed gene lists and conserved molecular signatures. Finally, we demonstrate how our Perturb-seq derived gene lists can be used to precisely infer changes in signalling pathway activation for in vivo and in situ samples. Our work enhances our understanding of signalling regulators and their targets, and lays a computational framework towards the data-driven inference of an ‘atlas’ of perturbation signatures.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
209,00 € per year
only 17,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
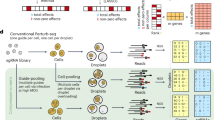


Data availability
Raw sequencing data that support the findings of this study have been deposited in GEO under accession code GSE281048. Processed data are available at Zenodo at https://zenodo.org/records/14518762 (ref. 119). Previously published datasets that were reanalysed here are available via either GEO (under accession codes GSE132080, GSE178429, GSE147405, GSE218033, GSE129390 and GSE169749) or the CZ CELLxGENE data portal117. Source data are provided with this paper.
Code availability
Software implementing our approach is freely available as an open-source R package Mixscale (https://github.com/satijalab/Mixscale). A vignette demonstrating the application of Mixscale is also available as an online resource (https://satijalab.github.io/Mixscale/).
References
Tang, F. et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015).
Picelli, S. et al. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098 (2013).
Buenrostro, J. D., Wu, B., Chang, H. Y. & Greenleaf, W. J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.1–21.29.9 (2015).
Farlik, M. et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 10, 1386–1397 (2015).
Stoeckius, M. et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017).
Rotem, A. et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 33, 1165–1172 (2015).
Mimitou, E. P. et al. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol. 39, 1246–1258 (2021).
Stubbington, M. J. T., Rozenblatt-Rosen, O., Regev, A. & Teichmann, S. A. Single-cell transcriptomics to explore the immune system in health and disease. Science 358, 58–63 (2017).
Van Hove, H. et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 22, 1021–1035 (2019).
Velmeshev, D. et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019).
Srivatsan, S. R. et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science 367, 45–51 (2020).
Mulder, K. et al. Cross-tissue single-cell landscape of human monocytes and macrophages in health and disease. Immunity 54, 1883–1900.e5 (2021).
Tabula Sapiens Consortium* et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014).
Wang, T., Wei, J. J., Sabatini, D. M. & Lander, E. S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014).
Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014).
Dixit, A. et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17 (2016).
Adamson, B. et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e21 (2016).
Jaitin, D. A. et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167, 1883–1896.e15 (2016).
Shifrut, E. et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e15 (2018).
Gasperini, M. et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 377–390.e19 (2019).
Replogle, J. M. et al. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28 (2022).
The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–D338 (2017).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503 (2020).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Morris, J. A. et al. Discovery of target genes and pathways at GWAS loci by pooled single-cell CRISPR screens. Science 380, eadh7699 (2023).
Yeo, N. C. et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 15, 611–616 (2018).
Börold, J. et al. BRD9 is a druggable component of interferon-stimulated gene expression and antiviral activity. EMBO Rep. 22, e52823 (2021).
Banno, T., Gazel, A. & Blumenberg, M. Effects of tumor necrosis factor-α (TNFα) in epidermal keratinocytes revealed using global transcriptional profiling. J. Biol. Chem. 279, 32633–32642 (2004).
Zeng, C.-M., Chen, Z. & Fu, L. Frizzled receptors as potential therapeutic targets in human cancers. Int. J. Mol. Sci. https://doi.org/10.3390/ijms19051543 (2018).
Sato, M. et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13, 539–548 (2000).
Kubiczkova, L., Sedlarikova, L., Hajek, R. & Sevcikova, S. TGF-β - an excellent servant but a bad master. J. Transl. Med. 10, 183 (2012).
Barkett, M. & Gilmore, T. D. Control of apoptosis by Rel/NF-κB transcription factors. Oncogene 18, 6910–6924 (1999).
Honda, K., Takaoka, A. & Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360 (2006).
Gordon, M. D. & Nusse, R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 281, 22429–22433 (2006).
Pico, A. R. et al. WikiPathways: pathway editing for the people. PLoS Biol. 6, e184 (2008).
Sanson, K. R. et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 9, 5416 (2018).
Balliu, B. et al. An integrated approach to identify environmental modulators of genetic risk factors for complex traits. Am. J. Hum. Genet. 108, 1866–1879 (2021).
Papalexi, E. et al. Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens. Nat. Genet. 53, 322–331 (2021).
Cook, D. P. & Vanderhyden, B. C. Context specificity of the EMT transcriptional response. Nat. Commun. 11, 2142 (2020).
Rhead, B. et al. Global expression and CpG methylation analysis of primary endothelial cells before and after TNFa stimulation reveals gene modules enriched in inflammatory and infectious diseases and associated DMRs. PLoS ONE 15, e0230884 (2020).
Hall, J. C. et al. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proc. Natl Acad. Sci. USA. 109, 17609–17614 (2012).
Kumaran Satyanarayanan, S. et al. IFN-β is a macrophage-derived effector cytokine facilitating the resolution of bacterial inflammation. Nat. Commun. 10, 1–16 (2019).
Tran, V. et al. High sensitivity single cell RNA sequencing with split pool barcoding. Preprint at bioRxiv https://doi.org/10.1101/2022.08.27.505512 (2022).
Almogy, G. et al. Cost-efficient whole genome-sequencing using novel mostly natural sequencing-by-synthesis chemistry and open fluidics platform. Preprint at bioRxiv https://doi.org/10.1101/2022.05.29.493900 (2022).
Squair, J. W. et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 12, 5692 (2021).
Wang, T., Li, B., Nelson, C. E. & Nabavi, S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinform. 20, 40 (2019).
Replogle, J. M. et al. Maximizing CRISPRi efficacy and accessibility with dual-sgRNA libraries and optimal effectors. eLife https://doi.org/10.7554/elife.81856 (2022).
Alerasool, N., Segal, D., Lee, H. & Taipale, M. An efficient KRAB ___domain for CRISPRi applications in human cells. Nat. Methods 17, 1093–1096 (2020).
Gilbert, L. A. et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661 (2014).
Xu, H. et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 25, 1147–1157 (2015).
Bosch, B. et al. Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell 184, 4579–4592.e24 (2021).
Hawkins, J. S. et al. Mismatch-CRISPRi reveals the co-varying expression-fitness relationships of essential genes in Escherichia coli and Bacillus subtilis. Cell Syst. 11, 523–535.e9 (2020).
Jost, M. et al. Titrating gene expression using libraries of systematically attenuated CRISPR guide RNAs. Nat. Biotechnol. 38, 355–364 (2020).
Simmons, S. K. et al. Mostly natural sequencing-by-synthesis for scRNA-seq using Ultima sequencing. Nat. Biotechnol. 41, 204–211 (2023).
Ivashkiv, L. B. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 18, 545–558 (2018).
Witten, D. M., Tibshirani, R. & Hastie, T. A penalized matrix decomposition, with applications to sparse principal components and canonical correlation analysis. Biostatistics 10, 515–534 (2009).
Jerby-Arnon, L. & Regev, A. DIALOGUE maps multicellular programs in tissue from single-cell or spatial transcriptomics data. Nat. Biotechnol. 40, 1467–1477 (2022).
François-Newton, V. et al. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLoS ONE 6, e22200 (2011).
Basters, A., Knobeloch, K.-P. & Fritz, G. USP18 - a multifunctional component in the interferon response. Biosci. Rep. https://doi.org/10.1042/bsr20180250 (2018).
Oshima, S. et al. Interferon regulatory factor 1 (IRF-1) and IRF-2 distinctively up-regulate gene expression and production of interleukin-7 in human intestinal epithelial cells. Mol. Cell. Biol. 24, 6298–6310 (2004).
Harada, H. et al. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell 58, 729–739 (1989).
Bien, J. & Tibshirani, R. Hierarchical clustering with prototypes via minimax linkage. J. Am. Stat. Assoc. 106, 1075–1084 (2011).
Zhu, Q. et al. Single cell multi-omics reveal intra-cell-line heterogeneity across human cancer cell lines. Nat. Commun. 14, 8170 (2023).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
Alsamman, K. & El-Masry, O. S. Interferon regulatory factor 1 inactivation in human cancer. Biosci. Rep. https://doi.org/10.1042/bsr20171672 (2018).
Pollaci, G. et al. Novel multifaceted roles for RNF213 protein. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23094492 (2022).
Grünvogel, O. et al. DDX60L is an interferon-stimulated gene product restricting hepatitis C virus replication in cell culture. J. Virol. 89, 10548–10568 (2015).
Karlsson, M. et al. A single-cell type transcriptomics map of human tissues. Sci Adv https://doi.org/10.1126/sciadv.abh2169 (2021).
Kang, H. M. et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 36, 89–94 (2018).
Kartha, V. K. et al. Functional inference of gene regulation using single-cell multi-omics. Cell Genom. https://doi.org/10.1016/j.xgen.2022.100166 (2022).
Rosain, J. et al. Human IRF1 governs macrophagic IFN-γ immunity to mycobacteria. Cell 186, 621–645.e33 (2023).
Irving, A. T. et al. Interferon regulatory factors IRF1 and IRF7 directly regulate gene expression in bats in response to viral infection. Cell Rep. 33, 108345 (2020).
Lei, X. et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 11, 3810 (2020).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Lee, J. S. et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci. Immunol. https://doi.org/10.1126/sciimmunol.abd1554 (2020).
Toro, A. et al. Pin-pointing the key hubs in the IFN-γ pathway responding to SARS-CoV-2 infection. Viruses https://doi.org/10.3390/v14102180 (2022).
Gadotti, A. C. et al. IFN-γ is an independent risk factor associated with mortality in patients with moderate and severe COVID-19 infection. Virus Res. 289, 198171 (2020).
Karki, R. et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184, 149–168.e17 (2021).
COvid-19 Multi-omics Blood ATlas (COMBAT) Consortium. A blood atlas of COVID-19 defines hallmarks of disease severity and specificity. Cell 185, 916–938.e58 (2022).
Kong, L. et al. The landscape of immune dysregulation in Crohn’s disease revealed through single-cell transcriptomic profiling in the ileum and colon. Immunity 56, 444–458.e5 (2023).
Parigi, S. M. et al. The spatial transcriptomic landscape of the healing mouse intestine following damage. Nat. Commun. 13, 828 (2022).
Schubert, M. et al. Perturbation-response genes reveal signaling footprints in cancer gene expression. Nat. Commun. 9, 20 (2018).
Beck, P. L. et al. Transforming growth factor-β mediates intestinal healing and susceptibility to injury in vitro and in vivo through epithelial cells. Am. J. Pathol. 162, 597–608 (2003).
Oshima, H. et al. Suppressing TGFβ signaling in regenerating epithelia in an inflammatory microenvironment is sufficient to cause invasive intestinal cancer. Cancer Res. 75, 766–776 (2015).
Penn, J. W., Grobbelaar, A. O. & Rolfe, K. J. The role of the TGF-β family in wound healing, burns and scarring: a review. Int. J. Burns Trauma 2, 18–28 (2012).
Datlinger, P. et al. Ultra-high-throughput single-cell RNA sequencing and perturbation screening with combinatorial fluidic indexing. Nat. Methods 18, 635–642 (2021).
Xu, Z., Sziraki, A., Lee, J., Zhou, W. & Cao, J. Dissecting key regulators of transcriptome kinetics through scalable single-cell RNA profiling of pooled CRISPR screens. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01948-9 (2023).
Lamb, J. et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006).
Calderon, D. et al. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet. 51, 1494–1505 (2019).
Urrutia, A. et al. Standardized whole-blood transcriptional profiling enables the deconvolution of complex induced immune responses. Cell Rep. 16, 2777–2791 (2016).
Jiang, P. et al. Systematic investigation of cytokine signaling activity at the tissue and single-cell levels. Nat. Methods 18, 1181–1191 (2021).
Cui, A. et al. Dictionary of immune responses to cytokines at single-cell resolution. Nature https://doi.org/10.1038/s41586-023-06816-9 (2023).
Goeva, A. et al. HiDDEN: a machine learning method for detection of disease-relevant populations in case-control single-cell transcriptomics data. Nat. Commun. 15, 1–15 (2024).
Song, B. et al. Decoding heterogenous single-cell perturbation responses. Nat. Cell Biol. https://doi.org/10.1038/s41556-025-01626-9 (2025).
Xie, Y. et al. Comparative analysis of single-cell RNA sequencing methods with and without sample multiplexing. Int. J. Mol. Sci. https://doi.org/10.3390/ijms25073828 (2024).
Rubin, A. J. et al. Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell 176, 361–376.e17 (2019).
Liscovitch-Brauer, N. et al. Profiling the genetic determinants of chromatin accessibility with scalable single-cell CRISPR screens. Nat. Biotechnol. 39, 1270–1277 (2021).
Pierce, S. E., Granja, J. M. & Greenleaf, W. J. High-throughput single-cell chromatin accessibility CRISPR screens enable unbiased identification of regulatory networks in cancer. Nat. Commun. 12, 2969 (2021).
Mimitou, E. P. et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 16, 409–412 (2019).
Wessels, H.-H. et al. Efficient combinatorial targeting of RNA transcripts in single cells with Cas13 RNA Perturb-seq. Nat. Methods 20, 86–94 (2023).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
McGinnis, C. S. et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nat. Methods 16, 619–626 (2019).
Satija, R. Barcoded plate-based single cell RNA-seq version 1. protocols.io https://doi.org/10.17504/protocols.io.nkgdctw (2018).
Ahlmann-Eltze, C. & Huber, W. glmGamPoi: fitting Gamma-Poisson generalized linear models on single cell count data. Bioinformatics 36, 5701–5702 (2021).
Mann, H. B. & Whitney, D. R. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Stat. 18, 50–60 (1947).
Marigorta, U. M., Rodríguez, J. A., Gibson, G. & Navarro, A. Replicability and prediction: lessons and challenges from GWAS. Trends Genet. 34, 504–517 (2018).
Seshadri, S. et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840 (2010).
Yasuda, K. et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 40, 1092–1097 (2008).
Hosack, D. A., Dennis, G. Jr, Sherman, B. T., Lane, H. C. & Lempicki, R. A. Identifying biological themes within lists of genes with EASE. Genome Biol. 4, R70 (2003).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Andreatta, M. & Carmona, S. J. UCell: robust and scalable single-cell gene signature scoring. Comput. Struct. Biotechnol. J. 19, 3796–3798 (2021).
CZI Single-Cell Biology Program et al. CZ CELL×GENE Discover: a single-cell data platform for scalable exploration, analysis and modeling of aggregated data. Nucleic Acids Res. 53, D886–D900 (2025).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Jiang, L. et al. Systematic reconstruction of molecular pathway signatures using scalable single-cell perturbation screens. Zenodo https://doi.org/10.5281/zenodo.14518762 (2024).
Acknowledgements
We thank all members of the Satija Laboratory at New York Genome Centre for useful discussion. We acknowledge the authors of the external datasets used in this study for making their valuable resources publicly available. This work was supported by the Chan Zuckerberg Initiative (EOSS5-0000000381 and HCA-A-1704-01895 to R.S.) and the National Institutes of Health (RM1HG011014-02 and 1OT2OD033760-01 and 5R01HD096770 to R.S.).
Author information
Authors and Affiliations
Contributions
L.J., C.D., E.P. and R.S. conceived the research. C.D., E.P., I.M., H.-H.W. and H.Y. performed experimental work. L.J. performed the computational work and developed the software tool with guidance from R.S. N.I., G.L.-Y. and D.L. performed the Ultima sequencing and generated the simulated paired-end fastq data. All authors participated in interpretation and in writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
In the past 3 years, R.S. has received compensation from Bristol Myers Squibb, ImmunAI, Resolve Biosciences, Nanostring, 10x Genomics, Parse Biosciences and Neptune Bio. R.S. and H.-H.W. are co-founders and equity holders of Neptune Bio. H.-H.W. has been an employee at Neptune Bio since August 2023. N.I., G.L.-Y. and D.L. are employees and shareholders of Ultima Genomics. E.P. has been an employee at Parse Biosciences since December 2021 and owns stock in the company. The other authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Optimization of a guide capture method compatible with Parse Biosciences Evercode Whole Transcriptome kits.
(a). Schematic of sgRNA capture, barcoding, and library preparation. Please see https://zenodo.org/records/14518762 for a full protocol. (b). Schematic showing the ___location of tested guide additive primer binding sites. (c). Primer efficiency for cDNA amplification, measured by qPCR with annealing temperature 65 °C. Guide additive with phosphorothioate (*) bonds was ultimately chosen. (d). Primer efficiency of chosen guide additive primer for cDNA amplification, measured by qPCR at different annealing temperatures. (e). Percentage of cells assigned to each guide classification when using the original Parse cDNA amplification annealing temperature (some cycles at 67 °C and some cycles at 65 °C) compared to our modified annealing temperature (all cycles at 65 °C). (f). RNA UMI counts and genes per cell when using the original Parse annealing temperature compared to our modified annealing temperature.
Extended Data Fig. 2 Validation of Mixscale perturbation scores across different sgRNAs and datasets.
(a) Scatter plots illustrating the relationship between the expression level of the perturbation targets (y axis) and the perturbation scores (x axis) in each cell. This plot is analogous to Fig. 2b but this time cells are stratified by their guide RNA identities instead. (b) Single-cell heatmap for STAT1 perturbation in three cell lines after IFNγ stimulation, split by sgRNA identities. (c) Comparison of Mixscale score and target gene expression estimated in an external CRISPRi dataset (Jost et al 2020 Nat. Biotechnol.). The figure displays the Mixscale score (y axis on the left) using black dots and the degree of knockdown of the target gene (y axis on the right) marked by red triangles. The x axis represents different sgRNAs, including the perfectly matched sgRNA (“_00”) and those with varying numbers of mismatched nucleotides. Plot shows that sgRNA that result in more effective knockdown also result in cells with higher Mixscale scores. (d) Comparison of Mixscale score and relative activity of sgRNAs. Similar plot as in (c), but instead the figure contrasts the Mixscale score (black dots) with the relative activity of the sgRNA (y axis on the right) marked by blue diamonds, a phenotypic measure of cellular growth defects measured from a viability screen. Plot shows that sgRNA with the highest phenotypic activity also yield cells with the highest Mixscale scores. Source numerical data are available in Source Data.
Extended Data Fig. 3 Mixscale perturbation scores correlate with target gene knockdown across varying perturbation strengths in IFNβ pathway.
Scatter plots illustrating the relationship between the expression level of the perturbation targets (y axis) and the perturbation scores (x axis) in each cell from the IFNβ pathway experiment. Plots are similar to Fig. 2b, but the perturbations are ordered based on the number of DEGs identified by standard Wilcoxon rank sum test (shown in parentheses for each panel) between the perturbed cells and non-targeting controls. Plots show that Mixscale perturbation scores correlate with the degree of knockdown for the target regulator for strong perturbations (with a large number of DEGs), and also for weaker perturbations (with fewer DEG). Source numerical data are available in Source Data.
Extended Data Fig. 4 False positive rate control and replication analysis of Mixscale differential expression testing.
(a). Comparison of false positive rates (FPRs) for the Mixscale weighted DE test (wmvReg), and performing the same test without weights (‘standard test’). FPRs are calculated based on alpha = 0.05, 0.01, and 0.005. Tests were performed on ‘null’ perturbations, generated by shuffling cell sgRNA labels after calculating Mixscale scores (Methods). Number of perturbations used for each pathway are: IFNβ (n = 52), IFNγ (n = 32), TGFβ (n = 15), and TNF (n = 29). (b). FPR calculations when shuffling is performed prior to calculating Mixscale scores. This situation is a conservative control. In a real dataset, the lack of an initially identified DEG set would abort the procedure (Methods) and assign uniform weights. Here, we force the assignment of Mixscale scores even though no DEG are identified after shuffling. Number of perturbations used for each pathway are: IFNβ (n = 52), IFNγ (n = 32), TGFβ (n = 15), and TNF (n = 29). (c). Boxplots for Mixscale DE test scores \({\chi }^{2}\) (= \({{Zscore}}^{2}\)) of the genes used in the calculation of these mis-specified scores, comparing methods with and without a leave-one-out (LOO) strategy (Methods). The red dashed line indicates the expected \({\chi }^{2}\) under the null. Number of observations (genes) used for each panel are: n10 = 3,206, n30 = 10,179, n50 = 16,975, and n100 = 33,956. In (a-c), each boxplot represents the distribution of FPR across all simulation replicates. The line inside each box indicates the median value, notches indicate the 95% confidence interval, central box indicates the interquartile range (IQR), whiskers indicate data up to 1.5 times the IQR, and all data points are shown as separate dots. (d). Replication rate of DE genes that were uniquely identified by Mixscale, compared to standard unweighted test. For example, in the IFNβ pathway dataset (Replicate 1), wmvReg and unweighted test uniquely identified 3,656 and 744 DEG, respectively, across regulators, and the plot shows the percentage that reproduce (P value cutoff for replication = 0.01) in the second replicate. (e) As in (d), but using Wilcoxon rank sum test instead of unweighted test. Source numerical data are available in Source Data.
Extended Data Fig. 5 Power analysis of differentially expressing genes and their reproducibility across different numbers of cell.
Analysis of how cell number affects DEG detection in IFNβ and IFNγ pathways. For each pathway, we downsampled our Perturb-seq data (replicate 1) to k cells (k = 30-1,000) per cell line per condition and performed differential expression analysis (Bonferroni-corrected P value < 0.05/30000). We conducted parallel analyses on replicate 2 (after downsampling to the same number of cells as in replicate 1) and calculated the percentage of DEGs from replicate 1 that were reproduced in replicate 2 (P < 0.01, see Methods). (a,c) Numbers and Reproducibility of DEGs with |fold change | ≥ 0.5 for IFNβ pathway (a) and IFNγ pathway (c). (b,d) Corresponding analyses for DEGs with |fold change | < 0.5 in IFNβ pathway (b) and IFNγ pathway (d).
Extended Data Fig. 6 Validation of Mixscale-specific differential expression genes using bulk RNA-seq data.
Comparison of DE genes identified by the Mixscale wmvReg test, and the standard unweighted test. (a-b) We generated independent bulk RNA-seq data of control and stimulated A549 cells (without genetic perturbations) for both IFNγ and TGFβ pathways, with three technical replicates (n = 3) for each condition. In our Perturb-seq data, DEG analysis was run on the IFNGR1 target gene (a), and the TGFBR1 target gene (b). Heatmaps showing the expression of shared genes (identified by both DE methods), and genes that were uniquely identified by Mixscale+wmvReg. For both pathways, <5 DEG were uniquely identified by the unweighted test. (c-d) Expression module score boxplots for both gene sets, quantifying the results in (a-b). These results indicate that the genes uniquely identified by Mixscale+wmvReg are indeed targets of the IFNγ (a,c) or TGFβ (b,d) pathway. Each boxplot represents the distribution of module scores across experimental replicates. The line inside each box indicates the median value, notches indicate the 95% confidence interval, central box indicates the interquartile range (IQR), whiskers indicate data up to 1.5 times the IQR, and all data points are shown as separate dots. Source numerical data are available in Source Data.
Extended Data Fig. 7 MultiCCA-derived perturbation programmes for IFNβ, TGFβ, and TNF pathways.
The first and second perturbation programmes for IFNβ, TGFβ, and TNF pathways, identified by MultiCCA. Each panel (a) IFNβ, (b) TGFβ, and (c) TNF, shows a heatmap where columns represent correlated perturbations within and across cell lines, and rows list the programme’s top 15 downregulated genes and top 5 upregulated genes. As in Fig. 3g, the colour gradient in the heatmap cells reflects the DE test Z-scores for each gene under each perturbation. See Table 3 for a complete lists of pathway programmes and the corresponding programme genes.
Extended Data Fig. 8 Comparative analysis of Perturb-seq and MSigDB pathway signatures.
(a). Venn diagrams showing the overlap between the MultiCCA programme 1 genes we identified and the MSigDB Hallmark gene lists for IFNγ, TGFβ, and TNF pathways (Methods). (b). Density plot for the log10(count per million) of MSigDB-unique, Perturb-seq-unique, and shared genes for each pathway, calculated in our Perturb-seq data (pseudobulk for all cells). The red dashed line indicates log10(CPM = 20). Plot shows that genes identified by MSigDB have a very different expression profile than those either unique identified by Perturb-seq or shared between the two databases (c). IFNβ module score comparing unstimulated and stimulated cells using the MSigDB-unique gene set. Plot shows that the MSigDB-unique gene sets effectively discriminate stimulated and control cells in only some cell types, in contrast with Perturb-seq gene sets in Fig. 4c. Source numerical data are available in Source Data.
Extended Data Fig. 9 Validation of Perturb-seq derived pathway signatures using cytokine-stimulated datasets.
(a-b). Evaluating complete pathway and pathway-exclusive gene sets. Plots are as in Fig. 4b, but run on datasets of cells that are stimulated with a single cytokine as a positive control. (a) shows the results from human CD14 monocytes stimulated with IFNγ, and (b) shows results from the DU145 cell line stimulated with TNF and results from the OVCA420 cells stimulated with TGFβ (Methods). In each case, our Perturb-seq pathway lists show enrichment, but there is also enriched signal for alternative pathways since pathway gene sets include overlapping genes. Once we restrict the analysis to pathway-exclusive gene sets, only the correct pathway exhibits evidence of enrichment. The enrichment tests used in (a-b) were two-sided Fisher’s exact tests. (c). The module score for IFNγ pathway genes, IRF1-associated genes, and IRF1-independent genes calculated in an IRF1-KO bat PakiT03 cell dataset (Methods). The IRF1-associated genes and IRF1-independent genes are identified using the IFNγ programme 1 and 2 in our Perturb-seq data (Methods). Source numerical data are available in Source Data.
Extended Data Fig. 10 Perturb-seq derived gene signatures show cell-type-specific enrichment in Crohn’s disease.
The gene set enrichment test for DEG identified for patients with Crohn’s disease (CD) in an external dataset (Kong et al 2023 Immunity) (Methods). The analysis includes inflamed and non-inflamed tissues from CD patients. Each row indicates a gene set from our Perturb-seq data, and each column indicates a cell type from which the DEGs are obtained. Two-sided Fisher’s exact tests were used for the enrichment test. The enrichment test odds ratio is represented by the size of the dot, and the enrichment test adjusted P value (after Benjamini–Hochberg correction) is represented by the gradient of the colour. Adjusted P-values less than 0.01 are labelled by asterisk. Source numerical data are available in Source Data.
Supplementary information
Supplementary Information
Supplementary Figs. 1–15.
Supplementary Tables
Supplementary Tables 1–5.
Source data
Source Data All Figures
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jiang, L., Dalgarno, C., Papalexi, E. et al. Systematic reconstruction of molecular pathway signatures using scalable single-cell perturbation screens. Nat Cell Biol 27, 505–517 (2025). https://doi.org/10.1038/s41556-025-01622-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41556-025-01622-z
This article is cited by
-
Decoding heterogeneous single-cell perturbation responses
Nature Cell Biology (2025)
