
Abstract
Up to 80% of rare disease patients remain undiagnosed after genomic sequencing1, with many probably involving pathogenic variants in yet to be discovered disease–gene associations. To search for such associations, we developed a rare variant gene burden analytical framework for Mendelian diseases, and applied it to protein-coding variants from whole-genome sequencing of 34,851 cases and their family members recruited to the 100,000 Genomes Project2. A total of 141 new associations were identified, including five for which independent disease–gene evidence was recently published. Following in silico triaging and clinical expert review, 69 associations were prioritized, of which 30 could be linked to existing experimental evidence. The five associations with strongest overall genetic and experimental evidence were monogenic diabetes with the known β cell regulator3,4 UNC13A, schizophrenia with GPR17, epilepsy with RBFOX3, Charcot–Marie–Tooth disease with ARPC3 and anterior segment ocular abnormalities with POMK. Further confirmation of these and other associations could lead to numerous diagnoses, highlighting the clinical impact of large-scale statistical approaches to rare disease–gene association discovery.
Similar content being viewed by others


Main
Rare diseases collectively affect 3.5% to 5.9% of people worldwide5. Despite advances in genomic sequencing, molecular diagnosis continues to elude 50% to 80% of patients presenting to genetic clinics1. Furthermore, fewer than half of the 10,000 rare Mendelian diseases in the Online Mendelian Inheritance in Man (OMIM) database6 have an established genetic basis. Diagnostic failure may arise because of a lack of routine screening for non-coding7 or structural variants1. However, it is likely that a substantial proportion of the pathogenic variants responsible for patients undiagnosed with rare disease (cases) reside in those yet to be discovered genes associated with (possibly very rare) disorders. The scale of rare disease sequencing studies, such as the Undiagnosed Disease Network8, Centers for Mendelian Genomics9, Deciphering Developmental Disorders10 and the 100,000 Genomes Project (100KGP)2, offers expanded opportunities to provide insight into pathogenic mechanisms of inherited disease, including the possibility of establishing disease–gene associations through case–control analyses, akin to methods used previously to identify common genetic variants influencing the risk of complex disorders. Such an approach provides much-needed power to identify genes harbouring rare pathogenic variants.
To identify disease-associated genes, we recently developed a framework that analyses rare protein-coding variants identified by the Exomiser variant prioritization tool11, in a preliminary version of the 100KGP data1, to conduct gene-based burden testing of single probands (first people in a family identified as affected by a rare genetic disease) and family members relative to control families. In silico triage in this previous study highlighted 22 new disease–gene associations, three of which have also been reported in independent studies12,13,14.
In this study, we have enhanced our gene burden analytical framework with a refined rare variant filtering and change of statistical modelling that are more tailored to Mendelian diseases and unbalanced case–control studies with rare events, extended it for generic application to any large-scale, rare disease sequencing cohorts, complemented it with visualization scripts and released it as an open-source R analytical framework called geneBurdenRD. In addition, we report on the application of the approach to a larger cohort from the final 100KGP data, including 34,851 families, 226 rare diseases and a starting pool of 4,643,230 rare candidate variants with improved in silico and added clinical expert triage of 69 probable new disease–gene associations.
Gene burden analytical framework
We have developed an open-source R framework (https://github.com/whri-phenogenomics/geneBurdenRD) allowing gene burden testing of variants in user-defined cases (sequenced probands affected by tested disease along with sequenced family members where available) versus controls from rare disease sequencing cohorts. The minimal input to the framework is (1) a file of rare, putative disease-causing variants obtained from merging and processing Exomiser output files for each of the cohort samples, (2) a file containing a label for each case–control association analysis to perform within the cohort and (3) a (set of) corresponding file(s) with user-defined identifiers and case–control assignment per sample. Cases and controls in a cohort could be defined in many ways, for example, by recruited disease category as we have done for the application to the 100KGP below, by specific phenotypic annotations or by phenotypic clustering. The 100KGP is perhaps atypical, compared with other projects, in having a virtual panel-based approach for all types of rare disease that required detailed inclusion and exclusion criteria for recruitment to specific disease categories, allowing accurate case–control definitions. Phenotype-based selection strategies would be required when this is not the case and can overcome false recruitment but will require comprehensive and accurate phenotyping for maximum effectiveness. As part of the variant quality control, the initial input of rare, putative disease-causing variants is further filtered down to remove possible false positive variant calls and/or relatively common variants in the project itself. Finally, for each case–control analysis, variants observed in cases are discarded if seen in at least one control to mimic a Mendelian, fully penetrant-like disease model. The framework then assesses false discovery rate (FDR)-adjusted disease–gene associations through a cohort allelic sums test (CAST) statistic used as covariate in a Firth’s logistic regression model (Methods). Genes are tested for enrichment in cases versus controls of rare, protein-coding, segregating variants that are (1) predicted loss-of-function (LoF), (2) highly predicted pathogenic (Exomiser variant score at least 0.8), (3) highly predicted pathogenic and present in a constrained coding region (CCR)15 or (4) de novo (restricted to only trios or larger families where de novo calling is possible and provided by the user) (Methods). As well as various output files annotating these case–control association tests, volcano plots are generated summarizing the FDR-adjusted P values of all the gene-based tests for each case–control association analysis, along with lollipop plots of the relevant variants in cases and controls and plots of the hierarchical distribution of the Human Phenotype Ontology (HPO) case annotations for individual disease–gene associations.
Application to 100KGP
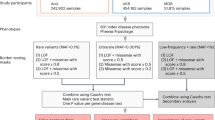
A rare variant gene burden analysis was performed on a cohort of 34,851 single probands and larger families (72,690 genomes), filtered down from an initial total of 35,548 (Methods), available from the 100KGP rare disease pilot and main programme (Data Release v.11) (Fig. 1 and Supplementary Table 1). The distribution of sex and genetically inferred ancestry in the overall cohort (Supplementary Table 1) is largely as expected from the reported ethnicity of the UK population (86% people of European descent, 8% people of Asian descent, 3% people of African/Caribbean descent, 2% mixed and 1% other in the 2011 census of England and Wales). A starting pool of 4,643,230 rare, protein-coding, segregating and most predicted pathogenic (per gene) variants for the analysis was derived by running Exomiser for each single proband and family and applying the initial variant quality control. Our pipeline was then used to detect statistically significant gene-based enrichment in relevant variant categories (predicted LoF, highly predicted pathogenic, highly predicted pathogenic in CCR regions, de novo) for ‘cases’ in each of 226 ‘specific diseases’ used for patient recruitment by the 100KGP1 versus ‘controls’ who were defined as probands from any other 20 broad ‘disease groups’ in the project (for example, intellectual disability cases were compared with all non-neurological probands as controls). A pairwise phenotypic comparison (Supplementary Fig. 1) highlights that the highest levels of similarity between samples are within these ‘specific disease’ categories, with extensive similarity often seen to other diseases in the same broad group (excluded in our control strategy), and no or more modest levels to the control disease categories (note that when combining all the control diseases, the median phenotypic similarity score between cases and controls was zero for all tested diseases). Applied cutoffs required at least five case probands per ‘specific disease’ and at least four probands (of which, at least two cases) with a relevant rare variant per disease–gene burden test. Overall, we performed 161 case–control gene burden analyses. Only gene-based enrichments that were more frequent in cases than controls (putative ‘disease-causing’ as opposed to ‘protective’) were considered in the statistical correction for multiple testing (51,899 association tests) (Fig. 1 and Supplementary Table 2; Methods).

Flowchart of the rare variant gene-based analytical framework, including triaging of results. aAfter sample quality control. bCAST statistic within Firth’s logistic regression model adjusted for sex, age, family size and inferred genetic ancestry. AD, autosomal dominant; oe_lof, observed/expected LoF variants.
We identified 165 previously known and 141 new potential disease–gene associations (Supplementary Table 3 and Extended Data Fig. 1), imposing a 0.5% FDR. At this threshold, we observed a reasonable recall (3.2%) and precision (47.4%) of currently known associations (using green genes in PanelApp16 only), a balanced number of known and new signals and no enrichment at all for synonymous variants (empirical negative control burden test). Not previously known (new) signals were initially defined, at the first round of analysis in March 2021, as having no documented evidence for an association within OMIM and absence from the ‘specific disease’ curated panel of high confidence (green) genes in PanelApp16. Enrichment of predicted LoF, highly predicted pathogenic, also in CCR and de novo variants was observed in 57%, 17%, 10% and 16% of known and 47%, 50%, 1% and 2% of new associations respectively, revealing discovery was driven mostly by predicted pathogenic, missense variants.
Of our 141 new signals, 5 have had interim independent supporting evidence (relevant OMIM entry and/or green gene in PanelApp) emerge since our initial assessment was performed in 2021: mitochondrial disorders with MORC2 (ref. 17); craniosynostosis with SOX6 (ref. 18); osteogenesis imperfecta with COPB2 (ref. 19); cystic kidney disease with IFT140 (ref. 20) and epidermolysis bullosa with TUFT1 (ref. 21).
The remaining potential associations were further filtered and prioritized to 69 of 136 (57%) by removing (1) those in which the gene was a non-protein-coding RNA (12/136); (2) those for signals driven by dominant, predicted LoF variants where the Genome Aggregation Database (gnomAD v.2.1.1)22 indicates there is no evidence for haplo-insufficiency (gnomAD observed/expected LoF at least 0.5) (19/136); or (3) those in which any of the cases driving the signals had already received an alternative genetic diagnosis (45/136) (Fig. 1 and Supplementary Table 4). It is possible but unlikely that criteria (3) will have removed genuine associations because of incorrect/partial diagnoses as the Genomics England diagnostic pipeline is fairly conservative, generally requiring predicted LoF, de novo or known pathogenic variants in well-established genes for a diagnosis. In comparison with the new association signals, 163 of 165 (99%) of signals from known disease genes passed criteria from (1) and (2). Variants responsible for the 69 associations were classified automatically according to ACMG using Exomiser, including whether predicted LoF variants fulfilled the latest recommendations for PVS1 (ref. 23) (Supplementary Table 5).
An extensive review of the literature as well as the phenotype evidence from Exomiser, in collaboration with members of the Genomics England Clinical Interpretation Partnerships (GeCIPs), was performed to identify supporting evidence from the biological function of each of the 69 genes, known disease associations or the phenotypes of the gene-deficient mouse and/or other animal models. In silico analyses were also undertaken to identify high quality StringDB24 protein–protein associations between the gene signal and any other genes known to be associated with the disease, or with highly specific expression in the most relevant tissue for the disease. This combined curation highlighted 30 associations supported by experimental evidence: 21 from literature curation of a gene function fitting the likely disease mechanism with further lines of evidence for many, 5 on the basis of mouse models and other evidence for some, and 4 on protein–protein evidence only (Fig. 1 and Supplementary Table 4 (highlighted in bold in the summary column)).
ClinGen25 has developed a robust set of criteria to assess the evidence for disease–gene associations and we applied these to our 69 associations. Evidence of causality was moderate for 27 associations and limited for the remainder (Fig. 1 and Supplementary Table 4). Of the 69 new associations, we chose candidates with previous functional data for the gene fitting the likely disease mechanism (Supplementary Table 4) and a ClinGen classification score of at least 8 to restrict to a manageable size of five candidates for further highlighting here.
Monogenic diabetes with UNC13A
We identified a dominant association (ClinGen moderate score of 9) between variants in UNC13A and specific disease ‘diabetes with additional phenotypes suggestive of a monogenic aetiology’. The association is driven by rare predicted LoF variants in two singleton cases with the only recorded phenotypes being diabetes mellitus in both and one further phenotype in one: p.Ala53Serfs*50 and p.Gly44* (adjusted P value, 0.0005; odds ratio, 329.8; 95% confidence interval, 58.2–1334.0; Fig. 2a and Supplementary Table 4). Both variants are absent from gnomAD v.4.1.0, classified as ‘likely pathogenic’ and predicted to undergo nonsense-mediated decay (NMD) (PVS1). The gene is depleted for rare LoF variants in gnomAD (observed/expected LoF = 0.09 (0.05–0.16) and probability of being LoF intolerant (pLI) = 1). UNC13A is a diacylglycerol and phorbol ester receptor gene with evidence for a role in the regulation of β cells3. Neonatal pancreatic β cells extracted from UNC13A-knockout mice and knock-in mice lacking the DAG binding ___domain show impaired second phase of insulin secretion in response to glucose stimulation4 and the heterozygous mouse knockout model shows impaired glucose tolerance26. In addition, co-expression network analysis of UNC13A and known monogenic diabetes genes from PanelApp shows the most significant enrichment in the pancreas Genotype Tissue Expression (GTEx) v.6 module (FDR-adjusted P value of 0.01), with UNC13A co-expressed with 6 out of 43 of the known genes. However, predicted LoF variants (three splice site, five stop gain or frameshift) were also seen in controls with no apparent history of diabetes indicating incomplete penetrance, later onset (year of birth of the two was 1973 and 1974 compared with 1956–2007 (mean 1980) for controls), or that the variants in controls are not genuinely LoF.

a, Location of rare, predicted pathogenic UNC13A variants associated with monogenic diabetes observed in two singleton cases. b, Location of rare, predicted pathogenic RBFOX3 variants associated with generalized epilepsies observed in two affected sisters and one singleton case; GTEx data showing specific brain expression. Het, heterozygous; hom ref, homozygous reference; TPM, transcripts per million.
Epilepsy with RBFOX3
We identified a dominant association (ClinGen moderate score of 11) between variants in RBFOX3 and specific disease ‘familial genetic generalized epilepsies’ (adjusted P value, 0.0023; odds ratio, 197.8; 95% confidence interval, 28.6–1,383.9; Fig. 2b and Supplementary Table 4). The association is driven by rare predicted pathogenic variants in two cases with seizure phenotypes: p.Asn105Asp in two affected sisters and p.Gln71* in a proband with further learning disability phenotypes; both variants absent from gnomAD v.4.1.0 and ClinVar and classified as variant of uncertain significance (VUS). A mouse model shows increased susceptibility to seizures27 and RBFOX3 is expressed specifically in the brain, particularly the cerebellum. A potential association between variants in RBFOX3 and epilepsy was published back in 201328 but no link is curated in OMIM or PanelApp and Gene Curation Coalition (GenCC) records limited evidence for the association. Our findings add strength to this association and a recent study has shown that RBFOX3 plays a critical role in the regulation of epilepsy and establishes it as a possible treatment path29.
Charcot–Marie–Tooth disease with ARPC3
We identified an association (ClinGen moderate score of 8) between variants in Actin Related Protein 2/3 Complex Subunit 3 (ARPC3) and specific disease ‘Charcot–Marie–Tooth (CMT) disease’. The association is driven by rare heterozygous variants in four cases: a p.Leu21Gln VUS observed in a duo and unrelated singleton case (allele frequency, 0.00001984 in gnomAD v.4.1.0 but in people of South Asian descent only), a p.Lys84dup disruptive inframe insertion VUS (absent from gnomAD v.4.1.0) in a singleton and a c.6 G>C VUS splice region variant (absent from gnomAD v.4.1.0) in a singleton (adjusted P value, 0.0015; odds ratio, 38.5; 95% confidence interval 9.6–144.6; Fig. 3a and Supplementary Table 4). The four cases show strong phenotypic similarity to each other (mean PhenoDigm score from pairwise, reciprocal, non-self hits was 0.84). Only 10 out of 1,000 randomly sampled CMT sets of the same size achieved the same mean score or higher, indicating the ARPC3 families with common features of distal upper and lower limb muscle weakness and peripheral axonal neuropathy are phenotypically distinct from the other CMT cases. Protein–protein associations are observed with known CMT genes: DNM2 and SYT2. Actin filaments play a key role in the neuronal cytoskeleton, the dysregulation of which is associated with various neurological conditions including CMT30, and ARPC3 may regulate dendritic spine morphology downstream of miR-29a/b31. An ArpC3 conditional knockout mice fails to ensheath axons32.

a, Location of rare, predicted pathogenic ARPC3 variants associated with CMT disease observed in three singleton cases and one proband not seen in the unaffected mother. b, Location of rare, predicted pathogenic POMK variants associated with corneal abnormalities observed in one singleton case and two trios with co-segregation from the affected mothers in the female probands; evidence of highly specific expression in the corneal epithelium from publicly available RNA-seq datasets (Methods). c, Location of rare, predicted pathogenic GPR17 variants associated with schizophrenia observed in two singleton cases. Comp het, compound heterozygous.
Corneal abnormalities with POMK
We identified a dominant association (ClinGen moderate score of 9) between variants in POMK and specific disease ‘corneal abnormalities’. The association is driven by rare, predicted pathogenic variants (two predicted LoF, one missense) in three cases with recorded phenotypes collectively suggestive of anterior segment dysgenesis (ASD) (adjusted P value, 0.0001; odds ratio, 151.5; 95% confidence interval, 37.6–473.3; Fig. 3b and Supplementary Table 4). ASD is a spectrum of developmental disorders affecting the anterior segment of the eye, often with incomplete penetrance and/or variable expressivity33. Co-segregation was apparent in two trios where a heterozygous, splice acceptor variant c.-21-1G>A (gnomAD v.4.1 allele frequency, 0.000011) and a heterozygous, frameshift stop gain variant p.Arg339* (gnomAD v.4.1 allele frequency, 0.000001) are inherited from the affected mothers in the female probands (Fig. 3b). Both predicted LoF variants were classified as VUS, with no NMD (PVS1) assignment. A further heterozygous missense variant p.Thr79Arg (gnomAD v.4.1 allele frequency, 0.000011) was observed in a singleton case and classified as ‘likely benign’. In the independent cohort described in Supplementary Table 4, one rare (gnomAD v.4.1 allele frequency, 0.000038), heterozygous missense variant p.Arg86His was identified in a mother and a son from a family from the Czech Republic diagnosed with ASD. POMK is involved in the presentation of the laminin-binding O-linked carbohydrate chain of alpha-dystroglycan, which forms transmembrane linkages between the extracellular matrix and the exoskeleton. Given the absence of corneal-specific expression data in the GTEx Project, we interrogated publicly available bulk RNA sequencing (RNA-seq) datasets34,35, which showed expression across all corneal cell types analysed, with highest amounts detected in the corneal epithelium (Fig. 3b). Bi-allelic (predicted LoF) variants in POMK are associated with autosomal recessive muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 12 (OMIM:615249)—a disease that also includes several ocular abnormalities (microphthalmia, buphthalmos, coloboma, retinal degeneration and cataract)—indicating that POMK plays a crucial role in ocular development36. Morpholino knockdown of the pomk gene in zebrafish has been reported to show several defects, including developmental ocular abnormalities36. Whether the identified rare variants here could induce ASD by POMK haplo-insufficiency, or by exerting dominant gain-of-function effects, deserves future investigation. With POMK apparently LoF-tolerant (pLI = 0) and ASD not having been reported in carriers of muscular dystrophy-dystroglycanopathy type A, 12-associated variants, the latter seems more likely.
Schizophrenia with GPR17
We identified an association (ClinGen moderate score of 9) between variants in GPR17 and specific disease ‘schizophrenia plus additional features’. The association is driven by rare predicted LoF variants in two singleton cases with schizophrenia and other psychiatric phenotypes: a p.Trp6* VUS (gnomAD v.4.1 allele frequency, 0.00018) (in compound-heterozygosity with p.Arg248Gln; allele frequency, 0.00032) and a heterozygous p.Glu129* VUS variant in the second case (allele frequency, 0.00001 in gnomAD v.4.1.0), both of which are predicted not to undergo NMD (adjusted P value, 0.0022; odds ratio, 189.7; 95% confidence interval, 29.1–882.0; Fig. 3c and Supplementary Table 4). GnomAD evidence (observed/expected LoF = 0.79 (0.45–1.46) and pLI = 0) does not support a haplo-insufficient mechanism for this gene. This may be explained by the variants acting in a recessive manner, or the high observed/expected LoF may represent the later onset, incomplete penetrance aspects of the disease and/or a polygenic mechanism of disease—the latter being probably given the phenotype. Direct protein–protein associations are recorded in StringDB for numerous other neurodevelopmental disorder genes: GABBR2, GNAI1, GNB1, GNB5, GPSM2, GRM1, PIK3CA, PIK3R2, PLCB1, PSAP, SPR and TRIO. Highly specific expression is observed in the brain, especially the cerebral cortex. Finally, GPR17 has been shown to regulate the oligodendrocyte differentiation and myelination that plays a role in several neurological diseases, including schizophrenia37.
Discussion
In this study, we have described a gene burden analysis of a large cohort of rare disease cases and identified 69 new disease–gene associations after triaging of the statistically significant signals. We highlight five with strong genetic and experimental evidence: monogenic diabetes associated with UNC13A, epilepsy with RBFOX3, CMT disease with ARPC3, anterior segment ocular abnormalities with POMK and schizophrenia with GPR17. However, further evidence is necessary before many of the new associations described here can be used clinically for diagnostics, counselling and management as a ClinGen classification of at least moderate evidence is required for inclusion in diagnostic genetic test panels38. For example, the addition of further strong functional study evidence would increase the score of the 42 limited ClinGen classification evidence candidates shown in Supplementary Table 4, such that they would be re-classified as moderate. We have submitted our evidence on the 69 associations to the GenCC database39. We are also pursuing the identification of variants in further independent cases through GeneMatcher (and linked Matchmaker Exchange nodes) for all our highlighted candidates, so far without success. Collection of more affected family members for cases in collaboration with the original recruiting clinicians could also raise the ClinGen category, although this is likely to be more difficult (for example, for a dominant association at least two large families with five affected members are needed to add one point of evidence).
Rare variant gene burden approaches have been used originally in the context of complex genetics where a steadily increased number of (typically) unrelated individuals, now reaching hundreds of thousands, are tested for genetic associations often across several quantitative and/or binary traits (genome-wide/phenome-wide association studies). In this context, variants that confer susceptibility to a certain disease are of interest and, also depending on the applied allele frequency threshold for variant filtering, people contributing several rare variants per gene are (typically) allowed in burden testing, this way increasing the chance to test variants with opposite effect (both protective and deleterious) in the same gene. Indeed, one of the main motivations behind the development of many of the existing gene burden tests beyond some basic implementations, for example, the CAST statistic used in our geneBurdenRD approach, is the need to tackle the presence of variants with different effect directions and/or effect sizes in the same gene40. Selection of the input rare variants is crucial in any gene burden approach and can affect the statistical power, and becomes paramount in the context of Mendelian diseases, where we are not after several susceptibility variants (in any effect direction), but rather a single putative disease-causative variant (or pair of variants in a compound heterozygous genotype). Therefore, a scenario where someone (whether case or control) contributes several rare genotypes to the same gene burden should not be contemplated. Our analytical framework (geneBurdenRD) is based on a convenient use of the established Exomiser variant prioritization tool for rare Mendelian diseases to perform the non-trivial step of variant annotation, scoring and segregation (using variant call format (VCF) files from single probands or family-based VCF files from nuclear families) and selection of the most predicted pathogenic, rare, segregating (putative disease-causing) variant(s) per proband/family and each gene (and each compatible mode of inheritance). geneBurdenRD also provides case–control variant count filtering that mimics a Mendelian fully penetrant-like disease model with variants seen in controls not included in the analysis.
Among state-of-the-art software for whole-genome regression modelling of genome-wide/phenome-wide association studies is REGENIE41, which has been shown to be substantially faster than other similar approaches such as FastGWA42, BOLT-LMM43 and SAIGE44. In addition to several statistical tests that are relevant mostly to complex genetics, such as single-variant, gene–gene and gene–environment interaction tests as well as conditional analyses, REGENIE and some of the other related software can perform various gene burden tests, for example, tailored tests to binary traits in both unbalanced and balanced case–control studies with rare variants, such as the saddle point approximation (SPA) approach45 and Firth’s logistic regression model46 (our statistical model of choice). Notably, both the SPA and the Firth’s correction have been shown to provide good control of Type 1 error, but the SPA approach implemented in SAIGE resulted in inflated effect-size estimates—a feature that was not observed with the Firth’s logistic regression in REGENIE41. Despite the ability to handle most commonly used input genetic and phenotypic files such as, for example, BGEN or PLINK/PLINK2, and tackle population structure and relatedness, REGENIE and similar approaches are not natively tailored to the analysis of rare, putative disease-causing, segregating variants within families for rare Mendelian diseases. The crucial and often cumbersome step of annotation, scoring and selection of the pool of relevant rare variants to test for gene burden is left to the user as a preliminary step with the suggestion to use external tools such as snpEFF or VEP. Our analytical framework geneBurdenRD overcomes this limitation and covers an important need for more tailored gene burden analytical tools for the analysis of rare Mendelian diseases, with approaches such as REGENIE remaining to be preferred for complex genetics analyses, across several quantitative and/or binary traits in (typically) unrelated people.
Although our approach applied to a large, rare disease cohort has successfully highlighted numerous known associations and indicated many previously unreported associations, it is not without limitations, pointing towards opportunities for future developments and re-analysis. For example, although we considered only single-nucleotide variants and small insertions or deletions in protein-coding genes in the current study, the inclusion of non-coding rare variation as well as structural variants may help unveil further molecular diagnoses and disease mechanisms. Despite the large-scale size of the 100KGP, 29 of the 226 diseases observed in this analysis have fewer than five cases and a further 36 did not pass our testing criterium of at least two same disease cases with relevant variants in a certain gene. This highlights the current limited power in discovering new (ultra-)rare monogenic disease associations and the need for even larger rare disease sequencing efforts. As the input to the statistical testing of each gene in our analytical framework is simply a matrix recording the presence or absence of a proband’s genotype passing each criterion for each case and control, one possibility to increase statistical power is federated analysis across rare disease sequencing projects as such (preferably, Exomiser-based) processed data should be sharable under most ethical and legal frameworks. Finally, whereas our focus was rare Mendelian, monogenic fully penetrant associations, further analytical developments and research are needed to uncover those missing genetic etiologies because of incomplete penetrance and/or variable expressivity as well as digenic and/or polygenic effects in rare diseases.
An alternative to our frequentist approach is offered by a Bayesian method for rare diseases called BeviMed47 (Bayesian evaluation of variant involvement in Mendelian disease), in which disease risk depends on the genotypes at rare variants in a locus, a latent mode of inheritance and a latent partition of variants into pathogenic and non-pathogenic subsets. BeviMed was used in a recent analysis of approximately the same cohort of rare disease patients in this study, where 241 known but only 19 new associations were found48. Indeed, 113 out of 165 of our known, and only 4 out of 141 of the potentially new (before triage), signals were also detected by BeviMed: TUFT1 associated with epidermolysis bullosa; SRP9 associated with ductal plate malformations, MORC2 associated with mitochondrial disorders and ARPC3 associated with CMT disease. Although a direct comparison is not straightforward because of the differences in the case–control selection strategies (the BeviMed study analysed 269 case sets as opposed to 161 that passed our testing criteria), let alone in the statistical approach, the relatively small overlap in signals highlights a possible complementarity of the two methods to discover new disease–gene associations from the same cohort.
There are 553 cases with no molecular diagnosis but with variant(s) contributing to one of the known disease–gene association signals that had not already been considered and classified as VUS or benign in the diagnostic report, giving an upper bound on the increase in diagnostic yield from review of these variants of 1.6% (553 of 34,851 cases analysed). Furthermore, 155 molecularly unsolved cases had a variant contributing to one of the 69 new associations giving an upper bound on the potential increase in diagnostic yield of 0.4% (155 of 34,851 cases analysed), if all genes were confirmed and the variants considered penetrant enough to be deemed pathogenic rather than just predictive. By making our analytical framework openly available for wider application to similar cohort data globally, we hope to substantially aid disease–gene discovery and new molecular diagnoses in rare Mendelian diseases in numerous other cohorts.
Methods
Rare disease genomes from the 100KGP
The Health Research Authority Research Ethics Committee East of England—Cambridge South (Ref. 14/EE/111) gave ethical approval for the 100KGP. Patients with rare diseases and affected and unaffected family members were enrolled to the 100KGP through 1 of the 13 NHS Genomic Medicine Centres across England, Northern Ireland, Scotland and Wales2. Consent was obtained from all participants to the 100KGP. The recruiting clinicians assigned each proband to a specific disease (according to a hierarchical disease classification available in the project described below) and provided patient’s phenotypic data according to the HPO49. An initial cohort of 74,061 genomes (35,548 single probands and larger families) from the rare disease pilot and main programme of the 100KGP (data release v.11) was available for analysis (March 2021). Genomes were sequenced using the TruSeq DNA polymerase-chain-reaction-free sample preparation kit (Illumina) on a HiSeq 2500 sequencer, which generates a mean depth of 32× (range, 27–54) and a depth greater than 15× for at least 95% of the reference human genome. Whole-genome sequencing reads were aligned to either the Genome Reference Consortium human genome build 37 (GRCh37) for the minority of earlier samples, or build 38 (GRCh38), with the use of Isaac Genome Alignment Software. Family-based variant calling of single-nucleotide variants and insertions or deletions for chromosomes 1 to 22, the X chromosome, and the mitochondrial genome (mean coverage, 2,814×; range, 142–16,581) was performed with the use of the Platypus variant caller50. Quality control performed by Genomics England highlighted that 81 of the probands had been recruited and sequenced twice and these duplicates were removed from our cohort. In addition, the required data for our Exomiser-based gene burden analysis, for example, recruited disease category and phenotypic terms, was not available for 16 families and these were also excluded from our cohort. The demographics of the cohort, presented in Supplementary Table 1, were obtained using Labkey in the Genomics England Research Environment. Genetic ancestry inference was performed by Genomics England by principal component analysis. A random forest model was subsequently trained to predict ancestry across five super-populations (European, African, Admixed American, South Asian, East Asian), with people assigned to ancestries on the basis of a probability threshold of greater than 0.8 (https://re-docs.genomicsengland.co.uk/ancestry_inference/).
Pool of rare, putative disease-causing variants for gene burden testing
The variant prioritization tool Exomiser11 (v.12.1.0 with default settings and latest 2007* (July 2020) databases) was then run on all available 35,451 single proband and family-based VCF files to obtain a pool of rare, protein-coding, segregating and most predicted pathogenic (per gene) variants to use in an rare variant gene-based burden testing analysis for the discovery of new rare Mendelian disease–gene associations as described below. For each proband/family and each gene, Exomiser selected a single configuration of contributing variants, that is, the most predicted (REVEL and MVP) pathogenic, rare (less than 0.1% autosomal/X-linked dominant or homozygous recessive, less than 2% autosomal/X-linked compound-heterozygote recessive; using publicly available sequencing datasets including gnomAD) protein-coding homozygous/heterozygous variant or compound-heterozygote variants that segregated with disease for each possible mode of inheritance. Coding variants (including canonical splice acceptor and donor and splice region (bases one to three of exon to three to eight of intron)) were selected by Exomiser by removing all those classified as FIVE_PRIME_UTR_EXON_VARIANT, FIVE_PRIME_UTR_INTRON_VARIANT, THREE_PRIME_UTR_EXON_VARIANT, THREE_PRIME_UTR_INTRON_VARIANT, NON_CODING_TRANSCRIPT_EXON_VARIANT, UPSTREAM_GENE_VARIANT, INTERGENIC_VARIANT, REGULATORY_REGION_VARIANT, CODING_TRANSCRIPT_INTRON_VARIANT, NON_CODING_TRANSCRIPT_INTRON_VARIANT and DOWNSTREAM_GENE_VARIANT. The Exomiser analysis did not return any candidate variants for 29 families, generally for larger families with several affected people where no rare, putative disease-causing variants remained after filtering, leading to an interim dataset size of 35,422 single probands and larger families. To control for false positive variant calls and/or relatively common variants in the project itself, we further discarded variants on the basis of how often they were observed in the Exomiser master dataset itself (frequency greater than 2% for variants in a compound-heterozygote genotype, greater than 0.2% for mitochondrial DNA genome variants, greater than 0.1% for heterozygote/homozygote variants). This led us to discard data from a further 41 families. Finally, potentially digenic probands with more than one recruited disease category were discarded from the analysis, leaving a total of 35,008 probands. As part of the sample quality control, kinship coefficients were used to control for cryptic relatedness. Genomics England provided kinship coefficients only for 29,180 of the 35,008 probands. Therefore, we calculated a genetic variant overlap measure (number of variants in common in the Exomiser results for two probands per total number of variants in the Exomiser results for the two probands) for all pairwise combinations of the 35,008 probands, and demonstrated that this was correlated strongly with the available kinship coefficients (Supplementary Fig. 2). Within each recruited disease category, we then identified genetically related probands (kinship coefficient greater than 0.088 corresponding to second-degree relatives or above, or an equivalent variant overlap score (0.1) threshold where kinship coefficient was not available). The proband in the pair with the most Exomiser results (less refined list of rare, putative disease-causing variants, usually from a smaller family size) was then dropped from all further analyses. This removed 157 probands/families, leading to a final input analysis dataset of 34,851 single probands and larger families (40,402 probands and affected family members and 32,288 unaffected family members) and 4,643,230 Exomiser-based candidate heterozygote/homozygote variants and compound-heterozygote genotypes (Supplementary Table 1 and Fig. 1). Furthermore, for each case–control analysis (see case–control definition below), a further variant quality control was applied with variants seen in at least one case (as heterozygote, homozygote or in a compound-heterozygote genotype) being discarded if seen in at least one control (as heterozygote/homozygote/compound-heterozygote, homozygote/compound-heterozygote, homozygote/compound-heterozygote, respectively) to mimic a Mendelian, fully penetrant-like disease model.
Exomiser-based rare variant gene burden testing for Mendelian diseases
A rare variant gene-based burden case–control analytical framework that exploits rare, putative disease-causing variants as annotated, filtered and scored by the variant prioritization tool Exomiser was used to identify new rare Mendelian disease–gene associations. The annotation of variants to genes comes from this Exomiser analysis using its default settings to identify the most damaging consequence to the set of Gencode-basic tagged Ensembl transcripts. The framework has been described previously1 and extended in this study (https://github.com/whri-phenogenomics/geneBurdenRD). Briefly, as to the application of the analytical framework to the rare disease component of the 100KGP, cases and controls were defined exploiting the hierarchical disease classification in the project itself where the recruiting clinicians assigned each proband to any of 228 ‘specific diseases’ (level 4); the ‘specific diseases’ are in turn grouped into less specific 91 ‘disease sub-groups’ (level 3), each of which corresponds to 1 of 20 broad ‘disease groups’ (level 2) (Supplementary Table 6). Two specific diseases (pontine tegmental cap dysplasia and childhood onset leukodystrophy) were never used for recruitment in the end, leaving a final set of 226 level 4 disease categories. A case set was then defined as all probands recruited under each of the 226 level 4 disease categories and its corresponding control set as all recruited probands except those under the level 2 category containing the specific level 4 disease, for example, hypertrophic cardiomyopathy cases were compared with all non-cardiovascular disorders probands as controls. As with the gene burden testing, the gene-based enrichment of variants in cases versus controls was quantified using the cohort allelic sums test (CAST)51 statistic under four proband genotype scenarios (irrespective of the mode of inheritance): (1) presence of at least one rare, predicted LoF variant; (2) presence of at least one rare, highly predicted pathogenic variant (Exomiser variant score of at least 0.8 (either predicted LoF or missense variants predicted to be pathogenic)); (3) presence of at least one rare, highly predicted pathogenic variant in a CCR and (4) presence of a rare, de novo variant (restricted to only trios or larger families where de novo calling is possible and provided by the user). These CCR regions were defined previously by looking for the absence of variation in gnomAD15 at various levels of certainty and in the application to the rare disease component of the 100KGP we used the 95% percentile download. Given that Exomiser selects by default a single configuration of ‘contributing’ variants for each proband/family, each gene and each possible mode of inheritance as compatible with available family-based data and we calculated the CAST statistic as the best observed irrespective of the mode of inheritance, the CAST statistic corresponds to a sum test statistic40. The gene burden association is then assessed either using a binary case–control status versus the CAST statistic in a right-tailed Fisher’s exact test as in the original implementation of the analytical framework1, or using the CAST statistic as a covariate in a Firth’s logistic regression model46 that is tailored to testing unbalanced case–control datasets with rare events. In the application to the rare disease component of the 100KGP, Firth’s logistic regression models were adjusted for age, sex, family size (single proband/duos/trios and larger families) and inferred genetic ancestry (Supplementary Table 1). To maintain statistical validity and power, the analysis was limited to those disease–gene associations where an arbitrary set of at least five cases exist for the specific disease tested and, for each of the four gene-based proband genotype scenarios above, where relevant variants in the gene were seen in at least four probands, of which at least two were cases (we would not follow up associations signals driven by single cases/families in the first instance). Only gene-based enrichments that were more frequent in cases than controls (putative disease-causing as opposed to protective) were considered in the statistical correction for multiple testing. The Benjamini and Hochberg method52 was used to correct for multiple testing; an overall FDR-adjusted P value threshold of 0.5% was used for claiming statistically significant disease–gene associations for further triaging.
Triaging
First in silico triage
The statistically significant associations were further filtered for those where (1) the gene was protein-coding as the Exomiser coding variant filtering settings also identified variants disrupting non-protein-coding RNA genes (gene type definitions from the human gene nomenclature committee (HGNC) website); (2) for dominant, LoF signals there was gnomAD evidence for haplo-insufficiency (gnomAD v.2.1.1 observed/expected LoF less than 0.5) and (3) none of the cases driving the signal were already assigned a molecular diagnosis in other genes as part of the 100KGP routine diagnostic pipeline.
Application of ClinGen framework for gene–disease validity
Classification of the disease–gene associations according to ClinGen criteria (https://clinicalgenome.org/docs/gene-disease-validity-standard-operating-procedures-version-10/) was applied using in silico approaches where possible. The case-level variant score was calculated from scoring and summing all case variants that support a particular mode of inheritance for a disease–gene association. LoF variants (stop gain, frameshift or splice acceptor/donor) scored 1.5 points or 2 if de novo, whereas others scored 0.1 points or 0.5 if de novo. A case–control study score of 5 points for an odds ratio greater than 5, 4 points for odds ratio greater than 3 or 3 points for odds ratio less than 3 was assigned. The larger of the case-level variant score or case–control study score was used as the genetic evidence score, capped at a maximum of 12 for those associations that had many supporting case variants. Experimental evidence categories were calculated using a variety of sources. Existing evidence for a gene function fitting the likely disease mechanism was assessed using PubMed searches using the disease and gene name and the background knowledge of the experts in the various disease-specific GeCIPs. Scores of 0, 1 or 2 were awarded depending on whether there was no, some tenuous or lots of evidence. Gene expression was assessed using GTEx Project data through the web portal of the Human Protein Atlas (https://www.proteinatlas.org/ (ref. 53); and/or publicly available relevant RNA-seq datasets34,35 processed with STAR v.2.7.6a and Salmon v.1.4.0, and a score of 0, 1 or 2 assigned for no, widespread or solely specific expression in the relevant disease tissue. Defaults of one point for protein–protein association evidence (high quality, direct experimental interactions scoring greater than 0.7 in StringDB with genes on the disease panel from PanelApp16) and 2 points for mouse/zebrafish evidence (phenotypic similarity as calculated by Exomiser between the patient’s phenotypes and the mouse/zebrafish phenotypes where the orthologous gene was disrupted) were used. The rounded sum of genetic and experimental evidence points was used to assign the final ClinGen classification of the evidence for the association as being limited (0.1–6 points), moderate (7–11 points) or strong (12–18 points). Definitive evidence for an association is considered to be a score of 12–18 as well as convincing replication of the result in more than two publications over more than 3 years. Therefore, none of our associations will be classified as definitive at this early stage.
Visual representation of variant ___location in lollipop plots
Visual representations of the variant locations in the protein were generated by extending the Mutplot software54. The x axis represents the amino acid chain and its annotated protein ___domain from UniProt. Each lolly indicates a variant by its protein change annotated on the MANE Select transcript or MANE Plus Clinical if a stronger impact is predicted (the transcript used is specified in the plot) and the frequency is shown on the y axis. Its shape indicates the genotype found in the proband. The colour indicates the type of variant and the variant’s functional annotation. If the variant has both a p. change annotation and a number in parenthesis it means that the original p. change was annotated on a different transcript and the amino acid position in parenthesis indicates the re-annotation on the selected transcript. If the only annotation available indicates a number in parenthesis it means that the variant was in the non-coding region for that transcript; therefore, the lolly was placed on the closest amino acid.
PhenoDigm patient similarity comparisons
During the assessment of some disease–gene associations, the phenotypic similarity between the probands driving the signal was calculated using their HPO term annotations and the Exomiser API to give a PhenoDigm55 score between 0 and 1. The mean of the pairwise, reciprocal, non-self hits was calculated and compared with those obtained from 1,000 iterations when the same number of probands was selected at random from the set of cases with that disease.
Co-expression network analysis
Co-expression network analysis of our candidate genes and known genes linked to the potentially associated disease (green genes in PanelApp v.1.120) was performed using GTEx v.6 tissue-specific modules and the CoExp tool accessible at https://rytenlab.com/coexp (ref. 56).
Peripheral blood mononuclear cell expression analysis
RNA-seq data from peripheral blood mononuclear cells collected from three volunteer donors was analysed (poly A-selected libraries, mean of two replicates untreated and two replicates treated with cycloheximide for 1 h to inhibit protein translation and mimic integrated stress response)57. In-house, R was used for DeSeq2 normalizations per library and calculation of the mean values for each transcript for the two replicate libraries per donor per condition. For global evaluations, across all three donors, the mean base value, log2 fold change post cycloheximide and Benjamini–Hochberg adjusted P value were then calculated.
Gene and variant look-up in independent rare disease cohorts
In a cohort of patients from the Irish Kidney Gene Project58 (278 cystic kidney disease and 141 chronic kidney disease cases), rare (gnomAD minor allele frequency less than 0.1%) LoF, missense, splicing or intronic variants were extracted for our new renal disease-associated genes. A further cohort of more than 3,000 Dutch renal patients was queried for likely pathogenic/pathogenic variants in those genes using the Alissa bioinformatics pipeline. Similarly, a sequencing cohort of 212 participants with inherited corneal diseases, recruited in the United Kingdom and Czech Republic and pre-screened for known genetic causes, was interrogated for any rare variants in the candidate gene POMK.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Access to the genetic and phenotypic data for the 100KGP participants is open to all through the Genomics England Research Environment (GeL RE) and by application at https://www.genomicsengland.co.uk/research/academic/join-gecip to become a member of the Genomics England Research Network. Multi-sample VCF files and PED files used to run Exomiser can be found under /genomes/analysis/rare_disease in the GeL RE file system. PanelApp gene panels and evidence of disease associations were obtained using the PanelApp API available at https://panelapp.genomicsengland.co.uk/api/docs/ (March 2021). Data used for UNC13A gene in Fig. 2, UniProt accession code Q9UPW8; data used for RBFOX3 gene in Fig. 2, UniProt accession code A6NFN3 and dbGaP accession code phs000424.v10.p2; data used for ARPC3 gene in Fig. 3: UniProt accession code, O15145; data used for POMK gene in Fig. 3, UniProt accession code Q9H5K3; GEO accession code GSE41616 and ENA accession code PRJEB1439;data for GPR17 gene in Fig. 3, UniProt accession code Q13304.
Code availability
The Exomiser-based rare variant gene burden R-based framework is available via Zenodo at https://doi.org/10.5281/zenodo.14500039 (ref. 59) as well as https://github.com/whri-phenogenomics/geneBurdenRD. The framework was developed using R v.4.2.1 (R packages: tidyverse v.2.0.0; data.table v.1.15.4; reshape2 v.1.4.4; biomaRt v.2.54.1; ggplot2 v.3.5.1; ggrepel v.0.9.5; httr v.1.4.7; drawProteins v.1.18.0; ensembldb v.2.22.20; AnnotationHub v.3.6.0; ontologyIndex v.2.12; ontologyPlot v.1.7). A script written in Perl (v.5.30.2) is provided to process the Exomiser output files and create the input file for the geneBurdenRD framework.
Change history
12 March 2025
Editor’s Note: Readers are alerted that technical concerns about the statistical approach have been raised, which could result in false positive gene associations being reported in this work. The editors are investigating these concerns and appropriate editorial action will be taken once this matter is resolved.
References
100,000 Genomes Project Pilot Investigators et al. 100,000 Genomes pilot on rare-disease diagnosis in health care—preliminary report. N. Engl. J. Med. 385, 1868–1880 (2021).
Turnbull, C. et al. The 100 000 Genomes Project: bringing whole genome sequencing to the NHS. BMJ 361, k1687 (2018).
Cataldo, L. R. et al. MAFA and MAFB regulate exocytosis-related genes in human β-cells. Acta Physiol. 234, e13761 (2022).
Kang, L. et al. Munc13-1 is required for the sustained release of insulin from pancreatic beta cells. Cell Metab. 3, 463–468 (2006).
Nguengang Wakap, S. et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur. J. Hum. Genet. 28, 165–173 (2020).
Amberger, J. S., Bocchini, C. A., Scott, A. F. & Hamosh, A. Omim.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 47, D1038–D1043 (2019).
Xiao, S. et al. Functional filter for whole-genome sequencing data identifies HHT and stress-associated non-coding SMAD4 polyadenylation site variants >5 kb from coding DNA. Am. J. Hum. Genet. 110, 1903–1918 (2023).
Splinter, K. et al. Effect of genetic diagnosis on patients with previously undiagnosed disease. N. Engl. J. Med. 379, 2131–2139 (2018).
Baxter, S. M. et al. Centers for Mendelian Genomics: a decade of facilitating gene discovery. Genet. Med. 24, 784–797 (2022).
Wright, C. F. et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 385, 1305–1314 (2015).
Bone, W. P. et al. Computational evaluation of exome sequence data using human and model organism phenotypes improves diagnostic efficiency. Genet. Med. 18, 608–617 (2016).
Farazi Fard, M. A. et al. Truncating mutations in UBAP1 cause hereditary spastic paraplegia. Am. J. Hum. Genet. 104, 767–773 (2019).
Wallmeier, J. et al. De novo mutations in FOXJ1 result in a motile ciliopathy with hydrocephalus and randomization of left/right body asymmetry. Am. J. Hum. Genet. 105, 1030–1039 (2019).
Cortese, A. et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 52, 473–481 (2020).
Havrilla, J. M., Pedersen, B. S., Layer, R. M. & Quinlan, A. R. A map of constrained coding regions in the human genome. Nat. Genet. 51, 88–95 (2019).
Martin, A. R. et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 51, 1560–1565 (2019).
Guillen Sacoto, M. J. et al. De novo variants in the ATPase module of MORC2 cause a neurodevelopmental disorder with growth retardation and variable craniofacial dysmorphism. Am. J. Hum. Genet. 107, 352–363 (2020).
Tolchin, D. et al. De novo SOX6 variants cause a neurodevelopmental syndrome associated with ADHD, craniosynostosis, and osteochondromas. Am. J. Hum. Genet. 106, 830–845 (2020).
Marom, R. et al. COPB2 loss of function causes a coatopathy with osteoporosis and developmental delay. Am. J. Hum. Genet. 108, 1710–1724 (2021).
Senum, S. R. et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am. J. Hum. Genet. 109, 136–156 (2022).
Jackson, A. et al. Biallelic TUFT1 variants cause woolly hair, superficial skin fragility and desmosomal defects. Br. J. Dermatol. 188, 75–83 (2023).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Singer-Berk, M. et al. Advanced variant classification framework reduces the false positive rate of predicted loss-of-function variants in population sequencing data. Am. J. Hum. Genet. 110, 1496–1508 (2023).
Szklarczyk, D. et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Rehm, H. L. et al. ClinGen—the clinical genome resource. N. Engl. J. Med. 372, 2235–2242 (2015).
Kwan, E. P. et al. Munc13-1 deficiency reduces insulin secretion and causes abnormal glucose tolerance. Diabetes 55, 1421–1429 (2006).
Wang, H.-Y. et al. RBFOX3/NeuN is required for hippocampal circuit balance and function. Sci. Rep. 5, 17383 (2015).
Lal, D. et al. RBFOX1 and RBFOX3 mutations in rolandic epilepsy. PLoS ONE 8, e73323 (2013).
Huang, D.-F. et al. Neuronal splicing regulator RBFOX3 mediates seizures via regulating Vamp1 expression preferentially in NPY-expressing GABAergic neurons. Proc. Natl Acad. Sci. USA 119, e2203632119 (2022).
Muñoz-Lasso, D. C., Romá-Mateo, C., Pallardó, F. V. & Gonzalez-Cabo, P. Much more than a scaffold: cytoskeletal proteins in neurological disorders. Cells 9, 358 (2020).
Lippi, G. et al. Targeting of the Arpc3 actin nucleation factor by miR-29a/b regulates dendritic spine morphology. J. Cell Biol. 194, 889–904 (2011).
Zuchero, J. B. et al. CNS myelin wrapping is driven by actin disassembly. Dev. Cell 34, 152–167 (2015).
Reis, L. M. et al. Comprehensive phenotypic and functional analysis of dominant and recessive FOXE3 alleles in ocular developmental disorders. Hum. Mol. Genet. 30, 1591–1606 (2021).
Chen, Y. et al. Identification of novel molecular markers through transcriptomic analysis in human fetal and adult corneal endothelial cells. Hum. Mol. Genet. 22, 1271–1279 (2013).
Bath, C. et al. Transcriptional dissection of human limbal niche compartments by massive parallel sequencing. PLoS ONE 8, e64244 (2013).
Di Costanzo, S. et al. POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations. Hum. Mol. Genet. 23, 5781–5792 (2014).
Lu, C. et al. G-protein-coupled receptor Gpr17 regulates oligodendrocyte differentiation in response to lysolecithin-induced demyelination. Sci. Rep. 8, 4502 (2018).
Bean, L. J. H. et al. Diagnostic gene sequencing panels: from design to report-a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 22, 453–461 (2020).
DiStefano, M. T. et al. The Gene Curation Coalition: a global effort to harmonize gene-disease evidence resources. Genet. Med. 24, 1732–1742 (2022).
Nicolae, D. L. Association tests for rare variants. Annu. Rev. Genomics Hum. Genet. 17, 117–130 (2016).
Mbatchou, J. et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 53, 1097–1103 (2021).
Jiang, L. et al. A resource-efficient tool for mixed model association analysis of large-scale data. Nat. Genet. 51, 1749–1755 (2019).
Loh, P.-R., Kichaev, G., Gazal, S., Schoech, A. P. & Price, A. L. Mixed-model association for biobank-scale datasets. Nat. Genet. 50, 906–908 (2018).
Zhou, W. et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341 (2018).
Kuonen, D. Miscellanea. Saddlepoint approximations for distributions of quadratic forms in normal variables. Biometrika 86, 929–935 (1999).
Firth, D. Bias reduction of maximum likelihood estimates. Biometrika 80, 27 (1993).
Greene, D., NIHR BioResource, Richardson, S. & Turro, E. A fast association test for identifying pathogenic variants involved in rare diseases. Am. J. Hum. Genet. 101, 104–114 (2017).
Greene, D. et al. Genetic association analysis of 77,539 genomes reveals rare disease etiologies. Nat. Med. 29, 679–688 (2023).
Köhler, S. et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 49, D1207–D1217 (2021).
Rimmer, A. et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 46, 912–918 (2014).
Morgenthaler, S. & Thilly, W. G. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST). Mutat. Res. 615, 28–56 (2007).
Benjamini, Y. & Hochberg, Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J. Educ. Behav. Stat. 25, 60 (2000).
Uhlén, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Zhang, W., Wang, C. & Zhang, X. Mutplot: an easy-to-use online tool for plotting complex mutation data with flexibility. PLoS ONE 14, e0215838 (2019).
Smedley, D. et al. PhenoDigm: analyzing curated annotations to associate animal models with human diseases. Database 2013, bat025 (2013).
García-Ruiz, S. et al. CoExp: a web tool for the exploitation of co-expression networks. Front. Genet. 12, 630187 (2021).
Li, D. et al. Endogenous plasma resuspension of peripheral blood mononuclear cells prevents preparative-associated stress that modifies polyA-enriched RNA responses to subsequent acute stressors. Cell Stress 11, 112–124 (2024).
Elhassan, E. A. E. et al. The utility of a genetic kidney disease clinic employing a broad range of genomic testing platforms: experience of the Irish Kidney Gene Project. J. Nephrol. 35, 1655–1665 (2022).
Cipriani, V., Vestito, L. & Smedley, D. whri-phenogenomics/geneBurdenRD: zenodo-v3 (zenodo-v3. Zenodo https://doi.org/10.5281/zenodo.14500039 (2024).
Acknowledgements
We thank L. Bottolo for statistical advice. This research was made possible through access to data in the National Genomic Research Library, which is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The National Genomic Research Library holds data provided by patients and collected by the NHS as part of their care and data collected as part of their participation in research. The National Genomic Research Library is supported by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also supported research infrastructure. P.L. was supported by GACR 24-10324S. This research was supported in part by Aligning Science Across Parkinson’s (grant nos. ASAP-000478 and ASAP-000509) through the Michael J. Fox Foundation for Parkinson’s Research (MJFF). K.A.B. and P. J. Conlon were supported by the Health Research Board and E.A.E.E. from Star MD. This is part of the NIHR Barts Biomedical Research Centre (Caulfield, Jones) portfolio of research. S.H. is a Wellcome Trust Clinical Career Development Fellow. W.G.N was supported by the Manchester NIHR BRC (NIHR 203308). C.L.S. was funded by the NIHR Imperial Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care. The analysis was supported by the NIH, National Institute of Child Health and Human Development through grant nos. 1R01HD103805-01 and 2UM1HG006370-11, and PhD funding from the UCLH NIHR BRC Deafness And Hearing Problems Theme (BRC392/HD/AS/110390). We thank M. Bitner–Glindzicz for her supervision.
Author information
Authors and Affiliations
Contributions
M.A.C., A.E.D. and P.L contributed to the analysis of the corneal patient cohort, evaluation of candidates and reviewing/editing the manuscript. A.M.v.E. and I.L. contributed to analysis of the Dutch cohort. S.E.F. contributed to the analysis of the Exeter diabetes patient cohort, evaluation of candidates and reviewing/editing the manuscript. P. J. Conlon, E.A.E.E. and O.T. contributed to the analysis of the Irish cohort. K.A.B. contributed to analysis of the Irish cohort, evaluation of candidates and reviewing/editing the manuscript. V.C., L.V. and D.S. developed the analysis pipeline, conducted analyses and cowrote the manuscript. M.C. contributed to the development of analysis and reviewing/editing the manuscript. P.N.R. contributed to the development of parts of the analysis pipeline. J.O.B.J. contributed to the development of parts of the analysis pipeline and reviewing/editing the manuscript. H.R.G., H.H., S.L., R.N., A.T.P., N.R., S.K.W. and S.L.Z. contributed to the evaluation of candidates. E.F.M., G.A., E.R.B., D.B., M.R.B., K.B., L.F.C., P. Chinnery, S.J.D., M.F., D.P.G., R.S.H., S.A.H., H.H.M., H.M., A.D.M., W.G.N., E.A.O., A.C.M.O., S.R., O.S.-A., J.A.S., J.C.T., I.T., A.T., J.S.W. and L.M.W. contributed to the evaluation of candidates and reviewing/editing the manuscript. S.G.-R. and A.R.H. contributed to the expression analysis. M.R. and C.L.S. contributed to the expression analysis, evaluation of candidates and reviewing/editing the manuscript. M.B., R.K., A.R.W. and C.T. contributed to the patient recruitment and phenotyping. C.G.C. and S.H. contributed to reviewing/editing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing interests: D.S. and M.C. were seconded to, and received salary from, Genomics England, a wholly owned Department of Health and Social Care company, from 2016 to 2018 and 2013 to 2021, respectively. E.A.O. has research funding from Kamari Pharma, Pavella Therapeutics, Unilever and the Leo Foundation unrelated to this work. She is CI for a trial for Kamari Pharma and performs consultancy for Kamari Pharma, Azitra and Palvella Therapeutics (all money goes to the university). S.L.Z. has provided consultancy services to Health Lumen. All other authors declare no competing interests.
Peer review
Peer review information
Nature thanks Heidi Rehm and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Rare disease gene discoveries from gene burden analysis of the 100,000 Genomes Project data.
The gene burden testing identified 306 disease-gene associations at 0.5% False Discovery Rate (FDR) including 141 potentially new. Initial triage of the new signals identified 69 signals for further investigation through in silico collection for additional evidence and clinical expert review. Statistical significance, expressed as -log10 of FDR-adjusted P value, is shown for each of the 306 disease–gene associations significant at 0.5% FDR, arranged by ‘disease group’. The 165 known associations are in green, 69 triaged new signals in blue, 5 new associations that had interim independent supporting evidence are in turquoise and the (new) triaged out signals in grey.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2.
Supplementary Table 1
Demographics of the cohort of 34,851 single probands and larger families recruited to the 100,000 Genomes Project used in the rare variant gene-burden analysis.
Supplementary Table 2
Gene burden summary statistics for the 51,899 association tests over 161 case-control analyses using Fisher’s test, Firth’s logistic regression and covariate-adjusted Firth’s logistic regression.
Supplementary Table 3
The 306 significant disease–gene identified associations by gene burden testing of the 100,000 Genomes Project data.
Supplementary Table 4
The 69 new, putative disease–gene associations discovered by gene burden testing of the 100,000 Genomes Project data.
Supplementary Table 5
ACMG classification of variants underlying each of the 69 new, putative disease-gene associations and corresponding ClinGen score.
Supplementary Table 6
Organisation of recruited (level 4) specific disease categories into (level 3) disease subgroups and (level 2) disease groups in the 100,000 Genomes Project.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cipriani, V., Vestito, L., Magavern, E.F. et al. Rare disease gene association discovery in the 100,000 Genomes Project. Nature (2025). https://doi.org/10.1038/s41586-025-08623-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41586-025-08623-w
