
Abstract
While genome-wide association studies are increasingly successful in discovering genomic loci associated with complex human traits and disorders, the biological interpretation of these findings remains challenging. Here we developed the GSA-MiXeR analytical tool for gene set analysis (GSA), which fits a model for the heritability of individual genes, accounting for linkage disequilibrium across variants and allowing the quantification of partitioned heritability and fold enrichment for small gene sets. We validated the method using extensive simulations and sensitivity analyses. When applied to a diverse selection of complex traits and disorders, including schizophrenia, GSA-MiXeR prioritizes gene sets with greater biological specificity compared to standard GSA approaches, implicating voltage-gated calcium channel function and dopaminergic signaling for schizophrenia. Such biologically relevant gene sets, often with fewer than ten genes, are more likely to provide insights into the pathobiology of complex diseases and highlight potential drug targets.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
209,00 € per year
only 17,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

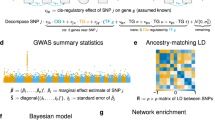
Data availability
The datasets analyzed in this study are available for download from the following URLs: Schizophrenia GWAS58 from PGC, https://pgc.unc.edu/for-researchers/download-results/; Molecular Signatures Database v7.5, https://www.gsea-msigdb.org/gsea/msigdb/; Synaptic Gene Ontologies 20210225 release, https://syngoportal.org/; functional categories (baselineLD_v2.2), https://alkesgroup.broadinstitute.org/LDSCORE/; 1000 Genomes Phase3 data, http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/. UKB, https://ams.ukbiobank.ac.uk/ams/; HRC Release 1.1, https://ega-archive.org/datasets/EGAD00001002729; TOP cohort, https://app.cristin.no/projects/show.jsf?id=2505365, and country-level GWAS summary statistics for PGC SCZ, https://pgc.unc.edu/for-researchers/data-access-committee/data-access-information/, have controlled data access. These data are not publicly available due to national data privacy regulations as they contain information that could compromise research participant privacy and/or consent. Statistical source data are provided for Figs. 1–4. The minimum dataset59 of this study is made publicly available and contains reference data formatted to GSA-MiXeR format, including definitions of genes, gene sets, functional categories and sharable nonsensitive data derived from UKB and HRC references. Source data are provided with this paper.
Code availability
The GSA-MiXeR software (v1.0.0), user tutorial and all codes required to process input data and generate the major results of this study are available from https://github.com/precimed/gsa-mixer60. Additionally, the following software packages were involved in performing data analysis: PLINK 1.90 build 20200616, https://www.cog-genomics.org/plink/1.9/; MAGMA v1.09b, https://ctg.cncr.nl/software/magma; LDAK v5.2, https://dougspeed.com/ldak-gbat/; KING v2.2.5, https://www.kingrelatedness.com/; flashpca 2.0, https://github.com/gabraham/flashpca; cleansumstats v1.6.0, https://github.com/BioPsyk/cleansumstats/; modified sLDSC v2023.05.15, https://github.com/ofrei/ldsc/tree/disable_jackknife; simu v0.9.3 pipeline to simulate synthetic GWAS from genotypes, https://github.com/precimed/simu; GWAS pipeline for UK Biobank v2, https://github.com/Nealelab/UK_Biobank_GWAS.
References
Sullivan, P. F. & Geschwind, D. H. Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell 177, 162–183 (2019).
de Leeuw, C. A., Neale, B. M., Heskes, T. & Posthuma, D. The statistical properties of gene-set analysis. Nat. Rev. Genet. 17, 353–364 (2016).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545 (2005).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Koopmans, F. et al. SynGO: an evidence-based, expert-curated knowledge base for the synapse. Neuron 103, 217–234.e4 (2019).
Hill, W. D. et al. A combined analysis of genetically correlated traits identifies 187 loci and a role for neurogenesis and myelination in intelligence. Mol. Psychiatry 24, 169–181 (2019).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Simillion, C., Liechti, R., Lischer, H. E. L., Ioannidis, V. & Bruggmann, R. Avoiding the pitfalls of gene set enrichment analysis with SetRank. BMC Bioinform. 18, 151 (2017).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Goeman, J. J. & Bühlmann, P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics 23, 980–987 (2007).
Tashman, K. C., Cui, R., O’Connor, L. J., Neale, B. M. & Finucane, H. K. Significance testing for small annotations in stratified LD-Score regression. Preprint at medRxiv https://doi.org/10.1101/2021.03.13.21249938 (2021).
Speed, D., Cai, N., Johnson, M. R., Nejentsev, S. & Balding, D. J. Reevaluation of SNP heritability in complex human traits. Nat. Genet. 49, 986–992 (2017).
Zabad, S., Ragsdale, A. P., Sun, R., Li, Y. & Gravel, S. Assumptions about frequency-dependent architectures of complex traits bias measures of functional enrichment. Genet. Epidemiol. 45, 621–632 (2021).
Frei, O. et al. Bivariate causal mixture model quantifies polygenic overlap between complex traits beyond genetic correlation. Nat. Commun. 10, 2417 (2019).
Holland, D. et al. Beyond SNP heritability: polygenicity and discoverability of phenotypes estimated with a univariate Gaussian mixture model. PLoS Genet. 16, e1008612 (2020).
Shadrin, A. A. et al. Phenotype-specific differences in polygenicity and effect size distribution across functional annotation categories revealed by AI-MiXeR. Bioinformatics 36, 4749–4756 (2020).
Holland, D. et al. The genetic architecture of human complex phenotypes is modulated by linkage disequilibrium and heterozygosity. Genetics 217, iyaa046 (2021).
Kingma, D.P. & Ba, J. L. Adam: a method for stochastic optimization. arXiv (2014).
Chen, J. et al. The trans-ancestral genomic architecture of glycemic traits. Nat. Genet. 53, 840–860 (2021).
Clarke, T. K. et al. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N=112 117). Mol. Psychiatry 22, 1376–1384 (2017).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261 (2017).
Evangelou, E. et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 50, 1412–1425 (2018).
Hautakangas, H. et al. Genome-wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype-specific risk alleles. Nat. Genet. 54, 152–160 (2022).
Mahajan, A. et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet. 50, 1505–1513 (2018).
Mishra, A. et al. Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 611, 115–123 (2022).
Okbay, A. et al. Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nat. Genet. 54, 437–449 (2022).
Savage, J. E. et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 50, 912–919 (2018).
Shah, S. et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat. Commun. 11, 163 (2020).
The, C.-H.G.I. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718 (2020).
Watanabe, K. et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 51, 1339–1348 (2019).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Wuttke, M. et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 51, 957–972 (2019).
Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in ~ 700000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649 (2018).
Smeland, O. B., Frei, O., Dale, A. M. & Andreassen, O. A. The polygenic architecture of schizophrenia—rethinking pathogenesis and nosology. Nat. Rev. Neurol. 16, 366–379 (2020).
Nakazawa, K. et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 62, 1574–1583 (2012).
Stedehouder, J. & Kushner, S. A. Myelination of parvalbumin interneurons: a parsimonious locus of pathophysiological convergence in schizophrenia. Mol. Psychiatry 22, 4–12 (2017).
Berrandou, T.-E., Balding, D. & Speed, D. LDAK-GBAT: fast and powerful gene-based association testing using summary statistics. Am. J. Hum. Genet. 110, 23–29 (2023).
Gazal, S. et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 49, 1421–1427 (2017).
Moon, A. L., Haan, N., Wilkinson, L. S., Thomas, K. L. & Hall, J. CACNA1C: association with psychiatric disorders, behavior, and neurogenesis. Schizophr. Bull. 44, 958–965 (2018).
Singh, T. et al. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 604, 509–516 (2022).
Howes, O. D. & Kapur, S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr. Bull. 35, 549–562 (2009).
Fusar-Poli, P. & Meyer-Lindenberg, A. Striatal presynaptic dopamine in schizophrenia, part II: meta-analysis of [18F/11C]-DOPA PET studies. Schizophr. Bull. 39, 33–42 (2013).
Huhn, M. et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: a systematic review and network meta-analysis. Lancet 394, 939–951 (2019).
Harrison, P. J. Schizophrenia susceptibility genes and neurodevelopment. Biol. Psychiatry 61, 1119–1120 (2007).
Burch, K. S. et al. Partitioning gene-level contributions to complex-trait heritability by allele frequency identifies disease-relevant genes. Am. J. Hum. Genet. 109, 692–709 (2022).
Yao, D. W., O’Connor, L. J., Price, A. L. & Gusev, A. Quantifying genetic effects on disease mediated by assayed gene expression levels. Nat. Genet. 52, 626–633 (2020).
Siewert-Rocks, K. M., Kim, S. S., Yao, D. W., Shi, H. & Price, A. L. Leveraging gene co-regulation to identify gene sets enriched for disease heritability. Am. J. Hum. Genet. 109, 393–404 (2022).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Zhu, X. & Stephens, M. Large-scale genome-wide enrichment analyses identify new trait-associated genes and pathways across 31 human phenotypes. Nat. Commun. 9, 4361 (2018).
Zhu, X., Duren, Z. & Wong, W. H. Modeling regulatory network topology improves genome-wide analyses of complex human traits. Nat. Commun. 12, 2851 (2021).
Shi, H., Kichaev, G. & Pasaniuc, B. Contrasting the genetic architecture of 30 complex traits from summary association data. Am. J. Hum. Genet. 99, 139–153 (2016).
Holland, D. et al. Estimating degree of polygenicity, causal effect size variance, and confounding bias in GWAS summary statistics. Preprint at bioRxiv (2017).
Storn, R. & Price, K. Differential evolution—a simple and efficient heuristic for global optimization over continuous spaces. J. Global Optim. 11, 341–359 (1997).
Nelder, J. A. & Mead, R. A simplex method for function minimization. Comput. J. 7, 308–313 (1965).
Brent, R. P. Algorithms for Minimization without Derivatives (Prentice-Hall, 1973).
Sullivan, P. Schizophrenia GWAS summary statistics. Figshare https://doi.org/10.6084/m9.figshare.19426775.v6 (2023).
Frei, O. Minimum dataset for GSA-MiXeR v1.0.0. Zenodo https://doi.org/10.5281/zenodo.10613336 (2024).
Frei, O. GSA-MiXeR v1.0.0 source code. Zenodo https://doi.org/10.5281/zenodo.10613326 (2024).
Acknowledgements
Funding was provided by the Research Council of Norway (RCN) #334920 to K.S.O.; Norwegian Health Association #22731 to S.B.; European Union’s Horizon 2020 (EU H2020) under the Marie Skłodowska-Curie Actions (Scientia fellowship) #801133 to N.P.; RCN #223273, #273291, #300309, #324252, #324499, #326813, South-Eastern Norway Regional Health Authority (HSØ) #2022073, The Kristian Gerhard Jebsen Stiftelsen #SKGJ-MED-021, EU H2020 grant #847776 (CoMorMent) and #964874 (RealMent), EEA and Norway grant #EEA-RO-NO-2018-0573, American National Institutes of Health (NIH) grant 5R01MH124839-02 (PGC4) to O.A.A.; NIH grants U24DA041123; R01AG076838; U24DA055330, OT2 HL161847 to A.M.D. This research has been conducted using the UK Biobank Resource under application number 27412. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), including Expanse and OASIS resources provided by San Diego Supercomputer Center at UC San Diego through allocation IBN200001. This work also used the TSD (Tjeneste for Sensitive Data) facilities, owned by the University of Oslo, operated and developed by the TSD service group at the University of Oslo, IT-Department (USIT, [email protected]), using resources provided by UNINETT Sigma2 – the National Infrastructure for High Performance Computing and Data Storage in Norway. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
O.F. conceived the study; O.F., A.A.S., N.P., S.B. and K.S.O. preprocessed the data; O.F., D.v.d.M, B.C.A. and E.H. performed all analyses, with conceptual input from G.H., A.A.S., C.d.L., D.P., W.C., D.H., O.B.S., O.A.A. and A.M.D.; O.F. and G.H. drafted the manuscript; all authors contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
A.M.D. was a Founder of and holds equity in CorTechs Labs, Inc., and serves on its Scientific Advisory Board. He is also a member of the Scientific Advisory Board of Human Longevity, Inc. (HLI), and the Mohn Medical Imaging and Visualization Centre in Bergen, Norway. He receives funding through a research agreement with General Electric Healthcare (GEHC). The terms of these arrangements have been reviewed and approved by the University of California, San Diego in accordance with its conflict-of-interest policies. O.A.A. has received speaker fees from Lundbeck, Janssen, Otsuka and Sunovion and is a consultant to Cortechs.ai unrelated to the topic of this study. C.d.L. is funded by Hoffman-La Roche. The remaining authors have no competing interest.
Peer review
Peer review information
Nature Genetics thanks Doug Speed, Martin Zhang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary methods, results and Figs. 1–11.
Supplementary Table 1–12
Supplementary Tables 1–12.
Source data
Source Data Fig. 1
Statistical source data for Fig. 1.
Source Data Fig. 2
Statistical source data for Fig. 2.
Source Data Fig. 3
Statistical source data for Fig. 3.
Source Data Fig. 4
Statistical source data for Fig. 4.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Frei, O., Hindley, G., Shadrin, A.A. et al. Improved functional mapping of complex trait heritability with GSA-MiXeR implicates biologically specific gene sets. Nat Genet 56, 1310–1318 (2024). https://doi.org/10.1038/s41588-024-01771-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-024-01771-1
This article is cited by
-
Elucidating shared genetic association between female body mass index and preeclampsia
Communications Biology (2025)
-
Genomics yields biological and phenotypic insights into bipolar disorder
Nature (2025)
-
A stratified treatment algorithm in psychiatry: a program on stratified pharmacogenomics in severe mental illness (Psych-STRATA): concept, objectives and methodologies of a multidisciplinary project funded by Horizon Europe
European Archives of Psychiatry and Clinical Neuroscience (2024)
