
Abstract
Haplotype-level allelic characterization facilitates research on the functional, evolutionary and breeding-related features of extremely large and complex plant genomes. We report a 21.7-Gb chromosome-level haplotype-resolved assembly in Pinus densiflora. We found genome rearrangements involving translocations and inversions between chromosomes 1 and 3 of Pinus species and a proliferation of specific long terminal repeat (LTR) retrotransposons (LTR-RTs) in P. densiflora. Evolutionary analyses illustrated that tandem and LTR-RT-mediated duplications led to an increment of transcription factor (TF) genes in P. densiflora. The haplotype sequence comparison showed allelic imbalances, including presence–absence variations of genes (PAV genes) and their functional contributions to flowering and abiotic stress-related traits in P. densiflora. Allele-aware resequencing analysis revealed PAV gene diversity across P. densiflora accessions. Our study provides insights into key mechanisms underlying the evolution of genome structure, LTR-RTs and TFs within the Pinus lineage as well as allelic imbalances and diversity across P. densiflora.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
209,00 € per year
only 17,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

Data availability
The genome assembly and annotation data for the haplotypes of P. densiflora and the genotype information generated from resequencing analysis of P. densiflora accessions have been deposited in figshare+ (https://doi.org/10.25452/figshare.plus.25546534)105. Resequencing data have been deposited at the NCBI SRA under BioProject accession number PRJNA1089250.
Code availability
The related code has been deposited in GitHub (https://github.com/minjeongjj/pinus_densiflora_haplotype_genome) and Zenodo (https://doi.org/10.5281/zenodo.12791823)106. All software used in this study is publicly available as described in Methods and Reporting Summary.
References
Garg, S. et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat. Biotechnol. 39, 309–312 (2021).
Guk, J. Y., Jang, M. J., Choi, J. W., Lee, Y. M. & Kim, S. De novo phasing resolves haplotype sequences in complex plant genomes. Plant Biotechnol. J. 20, 1031–1041 (2022).
Garg, S. Computational methods for chromosome-scale haplotype reconstruction. Genome Biol. 22, 101 (2021).
Kong, W., Wang, Y., Zhang, S., Yu, J. & Zhang, X. Recent advances in assembly of plant complex genomes. Genomics Proteomics Bioinformatics 21, 427–439 (2023).
Zhang, J. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 50, 1565–1573 (2018).
Sun, X. P. et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 52, 1423–1432 (2020).
Hasing, T. et al. A phased Vanilla planifolia genome enables genetic improvement of flavour and production. Nat. Food 1, 811–819 (2020).
Chen, H. et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 11, 2494–2504 (2020).
Sun, H. et al. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat. Genet. 54, 342–348 (2022).
Zhou, Q. et al. Haplotype-resolved genome analyses of a heterozygous diploid potato. Nat. Genet. 52, 1018–1023 (2020).
Farjon, A. & Filer, D. An Atlas of the World’s Conifers: an Analysis of their Distribution, Biogeography, Diversity and Conservation Status (Brill, 2013).
Jin, W.-T. et al. Phylogenomic and ecological analyses reveal the spatiotemporal evolution of global pines. Proc. Natl Acad. Sci. USA 118, e2022302118 (2021).
Moctezuma Lopez, G. & Flores, A. Economic importance of pine (Pinus spp.) as a natural resource in Mexico. Rev. Mex. Cienc. Forestales 11, 161–185 (2020).
Murthy, R., Dougherty, P. M., Zarnoch, S. J. & Allen, H. L. Effects of carbon dioxide, fertilization, and irrigation on photosynthetic capacity of loblolly pine trees. Tree Physiol. 16, 537–546 (1996).
Stevens, K. A. et al. Sequence of the sugar pine megagenome. Genetics 204, 1613–1626 (2016).
Pan, Y. et al. A large and persistent carbon sink in the world’s forests. Science 333, 988–993 (2011).
Kirst, M. et al. Apparent homology of expressed genes from wood-forming tissues of loblolly pine (Pinus taeda L.) with Arabidopsis thaliana. Proc. Natl Acad. Sci. USA 100, 7383–7388 (2003).
OBrien, I. E. W., Smith, D. R., Gardner, R. C. & Murray, B. G. Flow cytometric determination of genome size in Pinus. Plant Sci. 115, 91–99 (1996).
Neale, D. B. et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 15, R59–R71 (2014).
Niu, S. et al. The Chinese pine genome and methylome unveil key features of conifer evolution. Cell 185, 204–217 (2022).
Echt, C. S. et al. An annotated genetic map of loblolly pine based on microsatellite and cDNA markers. BMC Genet. 12, 17 (2011).
National Institute of Forest Science. SNP marker set for individual identification and population genetic analysis of Pinus densiflora and their use. KR patent 1020200045790 (2021).
Hirao, T. et al. Construction of genetic linkage map and identification of a novel major locus for resistance to pine wood nematode in Japanese black pine (Pinus thunbergii). BMC Plant Biol. 19, 424 (2019).
Liu, H. L. et al. The nearly complete genome of Ginkgo biloba illuminates gymnosperm evolution. Nat. Plants 7, 748–763 (2021).
Jin, J. et al. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 45, D1040–D1045 (2017).
Kim, S. et al. New reference genome sequences of hot pepper reveal the massive evolution of plant disease-resistance genes by retroduplication. Genome Biol. 18, 210–220 (2017).
Wang, P. et al. Genetic basis of high aroma and stress tolerance in the oolong tea cultivar genome. Hortic. Res. 8, 107 (2021).
Berardini, T. Z. et al. The Arabidopsis Information Resource: making and mining the ‘gold standard’ annotated reference plant genome. Genesis 53, 474–485 (2015).
Pascual, M. B., Canovas, F. M. & Avila, C. The NAC transcription factor family in maritime pine (Pinus pinaster): molecular regulation of two genes involved in stress responses. BMC Plant Biol. 15, 254 (2015).
Kang, M. et al. The C-___domain of the NAC transcription factor ANAC019 is necessary for pH-tuned DNA binding through a histidine switch in the N-___domain. Cell Rep. 22, 1141–1150 (2018).
Chakravarty, D. & Porter, L. L. AlphaFold2 fails to predict protein fold switching. Protein Sci. 31, e4353 (2022).
Millar, A. H., Carrie, C., Pogson, B. & Whelan, J. Exploring the function–___location nexus: using multiple lines of evidence in defining the subcellular ___location of plant proteins. Plant Cell 21, 1625–1631 (2009).
Han, J. et al. All-in-one: a robust fluorescent fusion protein vector toolbox for protein localization and BiFC analyses in plants. Plant Biotechnol. J. 20, 1098–1109 (2022).
Murcha, M. W., Kubiszewski-Jakubiak, S., Wang, Y. & Whelan, J. Evidence for interactions between the mitochondrial import apparatus and respiratory chain complexes via Tim21-like proteins in Arabidopsis. Front. Plant Sci. 5, 82 (2014).
Lister, R. et al. A transcriptomic and proteomic characterization of the Arabidopsis mitochondrial protein import apparatus and its response to mitochondrial dysfunction. Plant Physiol. 134, 777–789 (2004).
Samalova, M. et al. Hormone-regulated expansins: expression, localization, and cell wall biomechanics in Arabidopsis root growth. Plant Physiol. 194, 209–228 (2023).
Voith von Voithenberg, L. et al. A novel prokaryote-type ECF/ABC transporter module in chloroplast metal homeostasis. Front. Plant Sci. 10, 1264 (2019).
Li, W. et al. Plant pan-genomics: recent advances, new challenges, and roads ahead. J. Genet. Genomics 49, 833–846 (2022).
Leonard, A. S. et al. Structural variant-based pangenome construction has low sensitivity to variability of haplotype-resolved bovine assemblies. Nat. Commun. 13, 3012 (2022).
Karlgren, A., Gyllenstrand, N., Kallman, T. & Lagercrantz, U. Conserved function of core clock proteins in the gymnosperm Norway spruce (Picea abies L. Karst). PLoS ONE 8, e60110 (2013).
Lee, S., Hong, Y., Kwon, H. & Kim, Z. Population genetic studies on indigenous conifers in Korea. For. Sci. Technol. 2, 137–148 (2006).
Inglis, P. W., Pappas, M. C. R., Resende, L. V. & Grattapaglia, D. Fast and inexpensive protocols for consistent extraction of high quality DNA and RNA from challenging plant and fungal samples for high-throughput SNP genotyping and sequencing applications. PLoS ONE 13, e0206085 (2018).
Meng, L. & Feldman, L. A rapid TRIzol-based two-step method for DNA-free RNA extraction from Arabidopsis siliques and dry seeds. Biotechnol. J. 5, 183–186 (2010).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89 (2016).
Kim, S. et al. TGFam-Finder: a novel solution for target-gene family annotation in plants. New Phytol. 227, 1568–1581 (2020).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A. & Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121 (2013).
Gotz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435 (2008).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245–271 (2020).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Zhang, X. T. et al. Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant Camellia sinensis. Nat. Genet. 53, 1250–1274 (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457 (2020).
Yan, H., Bombarely, A. & Li, S. DeepTE: a computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 36, 4269–4275 (2020).
Tempel, S. Using and understanding RepeatMasker. Methods Mol. Biol. 859, 29–51 (2012).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18–31 (2008).
Steinbiss, S., Willhoeft, U., Gremme, G. & Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 37, 7002–7013 (2009).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49–e62 (2012).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Bailey, T. L. & Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36 (1994).
Bailey, T. L. & Gribskov, M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14, 48–54 (1998).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Loytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 1079, 155–170 (2014).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Jang, M. J., Hong, W. J., Park, Y. S., Jung, K. H. & Kim, S. Genomic basis of multiphase evolution driving divergent selection of zinc-finger homeodomain genes. Nucleic Acids Res. 51, 7424–7437 (2023).
Chae, G. Y., Hong, W. J., Jang, M. J., Jung, K. H. & Kim, S. Recurrent mutations promote widespread structural and functional divergence of MULE-derived genes in plants. Nucleic Acids Res. 49, 11765–11777 (2021).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Qiao, X. et al. Gene duplication and evolution in recurring polyploidization–diploidization cycles in plants. Genome Biol. 20, 38–60 (2019).
Marcais, G. et al. MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain. Fly 6, 80–92 (2012).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31–41 (2005).
Kim, M. H. et al. Wood transcriptome analysis of Pinus densiflora identifies genes critical for secondary cell wall formation and NAC transcription factors involved in tracheid formation. Tree Physiol. 41, 1289–1305 (2021).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–570 (2014).
Hyman, R. W., Jiang, H., Fukushima, M. & Davis, R. W. A direct comparison of the KB™ Basecaller and phred for identifying the bases from DNA sequencing using chain termination chemistry. BMC Res. Notes 3, 257 (2010).
Clough, S. J. & Bent, A. F. Floral dip: a simplified method for Agrobacterium‐mediated transformation of Arabidopsis thaliana. Plant J. 16, 735–743 (1998).
Norkunas, K., Harding, R., Dale, J. & Dugdale, B. Improving agroinfiltration-based transient gene expression in Nicotiana benthamiana. Plant Methods 14, 71 (2018).
Park, E., Lee, H. Y., Woo, J., Choi, D. & Dinesh-Kumar, S. P. Spatiotemporal monitoring of effectors via type III secretion using split fluorescent protein fragments. Plant Cell 29, 1571–1584 (2017).
Caplan, J. L. et al. Chloroplast stromules function during innate immunity. Dev. Cell 34, 45–57 (2015).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Tunyasuvunakool, K. et al. Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021).
Meng, E. C. et al. UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Zhang, Y. & Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33, 2302–2309 (2005).
Talenti, A. et al. A cattle graph genome incorporating global breed diversity. Nat. Commun. 13, 910 (2022).
Li, N. et al. Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species. Nat. Genet. 55, 852–860 (2023).
He, Q. et al. A graph-based genome and pan-genome variation of the model plant Setaria. Nat. Genet. 55, 1232–1242 (2023).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Gao, Y. et al. A pangenome reference of 36 Chinese populations. Nature 619, 112–121 (2023).
Hickey, G. et al. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 21, 35 (2020).
Siren, J. et al. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science 374, abg8871 (2021).
Garrison, E. et al. Building pangenome graphs. Preprint at bioRxiv https://doi.org/10.1101/2023.04.05.535718 (2023).
Graham, E. D., Heidelberg, J. F. & Tully, B. J. Potential for primary productivity in a globally-distributed bacterial phototroph. ISME J. 12, 1861–1866 (2018).
Kim, S., Jang, M.-J. & Cho, H. J. Chromosome-level haplotype-resolved genome assembly of Pinus densiflora. Figshare+ https://doi.org/10.25452/figshare.plus.25546534 (2024).
Jang, M.-J., Cho, H. J. & Kim, S. Code for chromosome-level haplotype-resolved genome assembly of Pinus densiflora (v1.0). Zenodo https://doi.org/10.5281/zenodo.12791823 (2024).
Acknowledgements
This study was supported by the Basic Science Research Program through a National Research Foundation of Korea grant funded by the Korean government (NRF-2022R1C1C1004918) to S.K., by the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, and Forestry through the Digital Breeding Transformation Technology Development Program funded by the Ministry of Agriculture, Food, and Rural Affairs (322075-3) to S.K., by a grant from the Korea Forest Service of the Korean government through the R&D Program for Forestry Technology (2014071H10-2022-AA04) to S.K. and E.-J.P. and by a National Institute of Forest Science grant (Forest Science Research project number FG0603-2021-01-2022) to E.-J.P. This research was supported in part by the Plant Biotic Interaction program of the National Science Foundation of the USA (NSF-IOS-2126256) to E.P. and J.W. and the intramural research program of the US Department of Agriculture, National Institute of Food and Agriculture Hatch Capacity (7000762) to E.P. We appreciate S. J. Lee of DNA Link, who provided support for genome sequencing analyses during this project.
Author information
Authors and Affiliations
Contributions
S.K. designed and organized the study as a lead contact. S.K. and E.-J.P. initiated the project. M.-J.J., H.J.C., Y.-S.P., S.-J.K., J.-W.C., G.Y.C., J.-Y.G. and S.K. performed data generation and/or bioinformatics analysis. M.-J.J., G.Y.C. and Y.-M.K. performed de novo genome assembly and annotation. H.J.C., Y.-S.P. and J.-W.C. performed TF gene analysis. M.-J.J. and H.J.C. performed haplotype variation and ortholog-based analysis. J.-W.C., J.-Y.G. and J.-H.K. performed the transcriptome analysis. M.-J.J., Y.-S.P., H.J.C. and M.-S.K. performed the resequencing analysis. D.Y.K., H.J.C., M.-J.J., S.-J.K. and S.-H.K. performed PAV and ASE validation. H.J.C. and E.-K.B. performed protein structure and abiotic stress analyses. E.-K.B., M.-J.K., H.L., K.-S.C., I.S.K., K.-S.K. and E.-J.P. prepared plant material, DNA and RNA samples and raw resequencing data. H.-Y.L., S.J., H.J., J.W., E.P. and D.C. performed ectopic expression and subcellular localization analyses. H.-Y.L. and E.P. designed, organized and drafted the initial manuscript of the ectopic expression and subcellular localization analyses. M.-J.J. and H.J.C. wrote the initial draft manuscript. S.K., M.-J.J. and H.J.C. reviewed and edited the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks De-Zhu Li, Yuanyuan Liu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
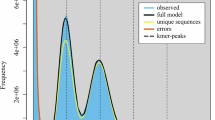
Extended Data Fig. 1 The k-mer analysis for genome size estimation and polymorphism of P. densiflora.
a, 21 k-mer depth distribution for genome size estimation. The x-axis indicates the k-mer depth and the y-axis indicates the frequency of k-mers. The dotted line represents the peak value. b, Haplotype-specific k-mer assembly spectrum (spectra-asm) plot from Merqury results. The graph shows k-mer proportion of haplotype-specific (red and blue), shared (green), and read-only (grey). The red and blue lines represent the evenly bisected the haplotype specific portion of k-mers, respectively.
Extended Data Fig. 2 Hi-C contact map for each of the 12 chromosomes in the P. densiflora genome.
a, Haplotype A (HA). b, Haplotype B (HB). The color scale from white to red indicates low to high contact probability.
Extended Data Fig. 3 Comparison of genomic analysis between P. densiflora and other Pinus species.
a, Genome-to-genome alignment of P. densiflora HA and other Pinus species. The color gradient from white to red represents gene-poor and gene-rich chromosome regions. The color gradient from white to green represents the increase in matched alignment length in each Pinus genome by the P. densiflora genome, while the height of the bar represents the alignment length in the P. densiflora genome by other Pinus genomes. The blue dotted line indicates the total proportion of matched genome sequences in the P. densiflora genome from other Pinus species. Asterisks (*) denote the top three highly duplicated regions in each chromosome of P. tabuliformis. b, Synteny comparison between P. densiflora HA and P. tabuliformis.
Extended Data Fig. 4 Genome rearrangements between chromosomes 1 and 3 in Pinus species.
Physical and genetic maps of Pinus species are depicted. The blocks (with a window size of 60 Mb) in the P. densiflora and P. tabuliformis genomes, mapped by representative markers of a, P. thunbergii, b, P. densiflora, and c, P. taeda are illustrated in grey color. LG, linkage group. In accordance with P. densiflora, the red and blue color for line, background, and marker represent chromosomes 1 and 3, respectively. Asterisks (*) denote the markers shown in Fig. 1c.
Extended Data Fig. 5 The correlation between the number of genes and LTR-RTs in the gypsy and copia subgroups on chromosomes of P. densiflora HA.
For each chromosome: a, upper correlation plots show LTR-RT subgroups that positively, negatively, or neutrally correlated with gene density and b, only neutrally correlated with gene density. The line colors indicate each LTR-RT subgroup. The number of genes (top) and LTR-RTs (bottom) were plotted as density within 30 Mb intervals. The color gradient from white to red, blue, and green represents increased number of genes, negatively correlated LTR-RTs with genes, and positively correlated LTR-RTs with genes, respectively.
Extended Data Fig. 6 Evolutionary analysis of Pinus and other plant species.
a, Evolution of gene families in 20 plant species. The numbers in blue and orange, separated by a slash, indicate the expanded and contracted gene families and rapidly evolved gene families, respectively (a two-sided P < 0.01) (left). On the right, the size and color of the circles indicate the number of rapid or not gene family expansion/contraction and gene gain/loss in each species. b, Domain repertoire of rapidly evolved genes in 4 Pinus species. The red, blue, orange, and green diamonds indicate 4, 3, 2, and individual Pinus species, respectively. c, Domain repertoire of genes in repeat regions of P. densiflora. d, Domain repertoire of rapidly evolved genes in individual Pinus species.
Extended Data Fig. 7 Enrichment analysis and phylogenetic relationships of TFs in Pinus, gymnosperm, and angiosperm.
a, The average number of TFs in Pinus (n = 4), gymnosperm (n = 7), and angiosperm (n = 9). Asterisks (***) denote a significance level of P < 0.001 based on a one-sided Fisher’s test. The individual data points are listed in Supplementary Table 3. Error bars indicate the standard error (SE). b, The phylogenetic relationships of TFs in 20 plant species. Dot colors on the nodes represents species, while the ring indicates subgroups.
Extended Data Fig. 8 Allelic imbalance of genic regions.
a, Genomic variations in genes including 2 kb upstream and downstream between P. densiflora HA and HB. The bar graphs show the total number of variations and overall number of genes containing SNPs, indels, and SVs in each genic region. b, Allelic gene categorization of P. densiflora. c, Domain repertoire of PAV and ASE genes. d, GO descriptions in biological process of PAV and ASE genes.
Extended Data Fig. 9 Validation of PAVs and ASEs in P. densiflora and characterization of Arabidopsis functional orthologous genes (FOGs).
a, Sequence validation of haplotype-specific presence of PAVs. b, Tissue abundant and haplotype unbalanced expression of ASEs in leaf, stem, and root. Asterisks (**) denote a significance level of P < 0.01 based on a one-sided unpaired Student’s t-test. At least two biological replicates are used. Error bars indicate the SE. The brown boxes and black lines indicate exons and introns, respectively. The red and blue backgrounds of amino acids indicate presence and absence, respectively. The hashtags (#) and asterisks (*) indicate frameshift mutation and stop codon, respectively. c, Arabidopsis FOGs annotated in P. densiflora HA and HB. d, Domain repertoire (left) and GO descriptions in biological process (right) of allele, PAV, and ASE genes. The pink, blue, and orange bars indicate allele, PAV, and ASE genes, respectively.
Extended Data Fig. 10 Genome-wide distribution for allele and PAV genes of P. densiflora and 30 wild accessions.
The outer track represents 12 chromosomes. The inner tracks represent gene density and PAV density for each accession.
Supplementary information
Supplementary Tables
Supplementary Tables 1–10.
Supplementary Data 1
Supplementary Data 1.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jang, MJ., Cho, H.J., Park, YS. et al. Haplotype-resolved genome assembly and resequencing analysis provide insights into genome evolution and allelic imbalance in Pinus densiflora. Nat Genet 56, 2551–2561 (2024). https://doi.org/10.1038/s41588-024-01944-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-024-01944-y
This article is cited by
-
Ovule development and pollen tube growth in Tsuga chinensis: insights into the evolution of siphonogamy
Plant Cell Reports (2025)
