
Abstract
Gene expression is controlled by dynamic localization of thousands of regulatory proteins to precise genomic regions. Understanding this cell type-specific process has been a longstanding goal yet remains challenging because DNA–protein mapping methods generally study one protein at a time. Here, to address this, we developed chromatin immunoprecipitation done in parallel (ChIP-DIP) to generate genome-wide maps of hundreds of diverse regulatory proteins in a single experiment. ChIP-DIP produces highly accurate maps within large pools (>160 proteins) for all classes of DNA-associated proteins, including modified histones, chromatin regulators and transcription factors and across multiple conditions simultaneously. First, we used ChIP-DIP to measure temporal chromatin dynamics in primary dendritic cells following LPS stimulation. Next, we explored quantitative combinations of histone modifications that define distinct classes of regulatory elements and characterized their functional activity in human and mouse cell lines. Overall, ChIP-DIP generates context-specific protein localization maps at consortium scale within any molecular biology laboratory and experimental system.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
209,00 € per year
only 17,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others

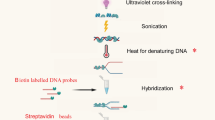

Data availability
All ChIP-DIP datasets generated in this study are available at GEO: GSE227773. Accession numbers for publicly available datasets used in this study are listed in Supplementary Methods.
Code availability
Publicly available software and packages were used in this study as indicated in Methods and Supplementary Methods. The original code for the ChIP-DIP pipeline is available on GitHub at https://github.com/GuttmanLab/chipdip-pipeline/tree/Paper (https://doi.org/10.5281/zenodo.13952458) (ref. 115).
References
Bednar, J. et al. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl Acad. Sci. USA 95, 14173–14178 (1998).
Jenuwein, T. & Allis, C. D. Translating the histone code. Science 293, 1074–1080 (2001).
Huang, H., Sabari, B. R., Garcia, B. A., Allis, C. D. & Zhao, Y. SnapShot: histone modifications. Cell 159, 458 (2014).
Tekel, S. J. & Haynes, K. A. Molecular structures guide the engineering of chromatin. Nucleic Acids Res. 45, 7555–7570 (2017).
Mashtalir, N. et al. Chromatin landscape signals differentially dictate the activities of mSWI/SNF family complexes. Science 373, 306–315 (2021).
He, S. et al. Structure of nucleosome-bound human BAF complex. Science 367, 875–881 (2020).
Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Barba-Aliaga, M., Alepuz, P. & Pérez-Ortín, J. E. Eukaryotic RNA polymerases: the many ways to transcribe a gene. Front. Mol. Biosci. 8, 663209 (2021).
Roeder, R. G. Role of general and gene-specific cofactors in the regulation of eukaryotic transcription. Cold Spring Harb. Symp. Quant. Biol. 63, 201–218 (1998).
Malik, S. & Roeder, R. G. Regulation of the RNA polymerase II pre-initiation complex by its associated coactivators. Nat. Rev. Genet. 24, 767–782 (2023).
Ho, L. & Crabtree, G. R. Chromatin remodelling during development. Nature 463, 474–484 (2010).
Johnson, D. S., Mortazavi, A., Myers, R. M. & Wold, B. Genome-wide mapping of in vivo protein–DNA interactions. Science 316, 1497–1502 (2007).
Mikkelsen, T. S. et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007).
Barski, A. et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007).
Robertson, G. et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4, 651–657 (2007).
He, Q., Johnston, J. & Zeitlinger, J. ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat. Biotechnol. 33, 395–401 (2015).
Serandour, A. A., Brown, G. D., Cohen, J. D. & Carroll, J. S. Development of an Illumina-based ChIP-exonuclease method provides insight into FoxA1–DNA binding properties. Genome Biol. 14, R147 (2013).
Tehranchi, A. K. et al. Pooled ChIP–seq links variation in transcription factor binding to complex disease risk. Cell 165, 730–741 (2016).
Aldridge, S. et al. AHT-ChIP–seq: a completely automated robotic protocol for high-throughput chromatin immunoprecipitation. Genome Biol. 14, R124 (2013).
Janssens, D. H. et al. Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia. Nat. Genet. 53, 1586–1596 (2021).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Skene, P. J., Henikoff, J. G. & Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 13, 1006–1019 (2018).
Lochs, S. J. A. et al. Combinatorial single-cell profiling of major chromatin types with MAbID. Nat. Methods 21, 72–82 (2024).
Gopalan, S., Wang, Y., Harper, N. W., Garber, M. & Fazzio, T. G. Simultaneous profiling of multiple chromatin proteins in the same cells. Mol. Cell 81, 4736–4746 (2021).
Gopalan, S. & Fazzio, T. G. Multi-CUT&Tag to simultaneously profile multiple chromatin factors. STAR Protoc. 3, 101100 (2022).
Kaya-Okur, H. S., Janssens, D. H., Henikoff, J. G., Ahmad, K. & Henikoff, S. Efficient low-cost chromatin profiling with CUT&Tag. Nat. Protoc. 15, 3264–3283 (2020).
Kong, N. R., Chai, L., Tenen, D. G. & Bassal, M. A. A modified CUT&RUN protocol and analysis pipeline to identify transcription factor binding sites in human cell lines. STAR Protoc. 2, 100750 (2021).
Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
PsychENCODE Consortium et al. The PsychENCODE project. Nat. Neurosci. 18, 1707–1712 (2015).
The Immunological Genome Project Consortium et al. The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094 (2008).
Partridge, E. C. et al. Occupancy maps of 208 chromatin-associated proteins in one human cell type. Nature 583, 720–728 (2020).
He, Y. et al. Spatiotemporal DNA methylome dynamics of the developing mouse fetus. Nature 583, 752–759 (2020).
Sisu, C. et al. Transcriptional activity and strain-specific history of mouse pseudogenes. Nat. Commun. 11, 3695 (2020).
Chasman, D. & Roy, S. Inference of cell type specific regulatory networks on mammalian lineages. Curr. Opin. Syst. Biol. 2, 130–139 (2017).
Ota, M. et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell 184, 3006–3021 (2021).
Madhani, H. D. et al. Epigenomics: a roadmap, but to where? Science 322, 43–44 (2008).
Kidder, B. L., Hu, G. & Zhao, K. ChIP–seq: technical considerations for obtaining high-quality data. Nat. Immunol. 12, 918–922 (2011).
Quinodoz, S. A. et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757 (2018).
Quinodoz, S. A. et al. RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790 (2021).
Quinodoz, S. A. et al. SPRITE: a genome-wide method for mapping higher-order 3D interactions in the nucleus using combinatorial split-and-pool barcoding. Nat. Protoc. 17, 36–75 (2022).
Kim, S., Yu, N.-K. & Kaang, B.-K. CTCF as a multifunctional protein in genome regulation and gene expression. Exp. Mol. Med. 47, e166 (2015).
Kouzarides, T. Chromatin modifications and their function. Cell 128, 693–705 (2007).
Girbig, M., Misiaszek, A. D. & Müller, C. W. Structural insights into nuclear transcription by eukaryotic DNA-dependent RNA polymerases. Nat. Rev. Mol. Cell Biol. 23, 603–622 (2022).
Abascal, F. et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Adli, M., Zhu, J. & Bernstein, B. E. Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nat. Methods 7, 615–618 (2010).
Karimzadeh, M. & Hoffman, M. M. Virtual ChIP–seq: predicting transcription factor binding by learning from the transcriptome. Genome Biol. 23, 126 (2022).
Ernst, J. & Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 12, 2478–2492 (2017).
Spicuglia, S. & Vanhille, L. Chromatin signatures of active enhancers. Nucleus 3, 126–131 (2012).
Steger, D. J. et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 28, 2825–2839 (2008).
Gates, L. A., Foulds, C. E. & O’Malley, B. W. Histone marks in the ‘driver’s seat’: functional roles in steering the transcription cycle. Trends Biochem. Sci. 42, 977–989 (2017).
Karmodiya, K., Krebs, A. R., Oulad-Abdelghani, M., Kimura, H. & Tora, L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genomics 13, 424 (2012).
Chen, Z., Djekidel, M. N. & Zhang, Y. Distinct dynamics and functions of H2AK119ub1 and H3K27me3 in mouse preimplantation embryos. Nat. Genet. 53, 551–563 (2021).
Saksouk, N., Simboeck, E. & Déjardin, J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin 8, 3 (2015).
Chen, T. & Dent, S. Y. R. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat. Rev. Genet. 15, 93–106 (2014).
Kirtana, R., Manna, S. & Patra, S. K. Molecular mechanisms of KDM5A in cellular functions: facets during development and disease. Exp. Cell Res. 396, 112314 (2020).
Shilatifard, A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr. Opin. Cell Biol. 20, 341–348 (2008).
Geng, Z. & Gao, Z. Mammalian PRC1 complexes: compositional complexity and diverse molecular mechanisms. Int. J. Mol. Sci. 21, 8594 (2020).
Mierlo, G., van, Veenstra, G. J. C., Vermeulen, M. & Marks, H. The complexity of PRC2 subcomplexes. Trends Cell Biol. 29, 660–671 (2019).
Bosch-Presegué, L. et al. Mammalian HP1 isoforms have specific roles in heterochromatin structure and organization. Cell Rep. 21, 2048–2057 (2017).
Mazzocca, M., Colombo, E., Callegari, A. & Mazza, D. Transcription factor binding kinetics and transcriptional bursting: what do we really know? Curr. Opin. Struct. Biol. 71, 239–248 (2021).
Bartman, C. R. et al. Transcriptional burst initiation and polymerase pause release are key control points of transcriptional regulation. Mol. Cell 73, 519–532 (2019).
Rada-Iglesias, A. et al. Whole-genome maps of USF1 and USF2 binding and histone H3 acetylation reveal new aspects of promoter structure and candidate genes for common human disorders. Genome Res. 18, 380–392 (2008).
O’Connor, L., Gilmour, J. & Bonifer, C. The role of the ubiquitously expressed transcription factor Sp1 in tissue-specific transcriptional regulation and in disease. Yale J. Biol. Med. 89, 513–525 (2016).
Li, Z., Cogswell, M., Hixson, K., Brooks-Kayal, A. R. & Russek, S. J. Nuclear respiratory factor 1 (NRF-1) controls the activity dependent transcription of the GABA-A receptor β1 subunit gene in neurons. Front. Mol. Neurosci. 11, 285 (2018).
Horn, H. F. & Vousden, K. H. Coping with stress: multiple ways to activate p53. Oncogene 26, 1306–1316 (2007).
Fischer, M. Census and evaluation of p53 target genes. Oncogene 36, 3943–3956 (2017).
Akberdin, I. R. et al. Pluripotency gene network dynamics: system views from parametric analysis. PLoS ONE 13, e0194464 (2018).
Reith, W. et al. MHC class II regulatory factor RFX has a novel DNA-binding ___domain and a functionally independent dimerization ___domain. Genes Dev. 4, 1528–1540 (1990).
Brivanlou, A. H. & Darnell, J. E. Signal transduction and the control of gene expression. Science 295, 813–818 (2002).
Satoh, J., Kawana, N. & Yamamoto, Y. Pathway analysis of ChIP–seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul. Syst. Biol. 7, GRSB.S13204 (2013).
Qi, B., Newcomer, R. & Sang, Q.-X. ADAM19/adamalysin 19 structure, function, and role as a putative target in tumors and inflammatory diseases. Curr. Pharm. Des. 15, 2336–2348 (2009).
Schoch, S., Cibelli, G. & Thiel, G. Neuron-specific gene expression of synapsin I. Major role of a negative regulatory mechanism. J. Biol. Chem. 271, 3317–3323 (1996).
Martin, D. & Grapin-Botton, A. The importance of REST for development and function of beta cells. Front. Cell Dev. Biol. 5, 12 (2017).
Bao, F., LoVerso, P. R., Fisk, J. N., Zhurkin, V. B. & Cui, F. p53 binding sites in normal and cancer cells are characterized by distinct chromatin context. Cell Cycle 16, 2073–2085 (2017).
Otto, S. J. et al. A new binding motif for the transcriptional repressor REST uncovers large gene networks devoted to neuronal functions. J. Neurosci. 27, 6729–6739 (2007).
Garber, M. et al. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol. Cell 47, 810–822 (2012).
Ernst, J. & Kellis, M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 28, 817–825 (2010).
Bernstein, B. E. et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006).
Wang, H. et al. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature 615, 339–348 (2023).
Bilodeau, S., Kagey, M. H., Frampton, G. M., Rahl, P. B. & Young, R. A. SetDB1 contributes to repression of genes encoding developmental regulators and maintenance of ES cell state. Genes Dev. 23, 2484–2489 (2009).
Zentner, G. E. & Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 20, 259–266 (2013).
Giaimo, B. D. et al. Histone variant H2A.Z deposition and acetylation directs the canonical Notch signaling response. Nucleic Acids Res. 46, 8197–8215 (2018).
Giaimo, B. D., Ferrante, F., Herchenröther, A., Hake, S. B. & Borggrefe, T. The histone variant H2A.Z in gene regulation. Epigenetics Chromatin 12, 37 (2019).
Gévry, N., Chan, H. M., Laflamme, L., Livingston, D. M. & Gaudreau, L. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 21, 1869–1881 (2007).
Gévry, N. et al. Histone H2A.Z is essential for estrogen receptor signaling. Genes Dev. 23, 1522–1533 (2009).
Akerberg, B. N. et al. A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat. Commun. 10, 4907 (2019).
Currey, L., Thor, S. & Piper, M. TEAD family transcription factors in development and disease. Development 148, dev196675 (2021).
Meers, M. P., Llagas, G., Janssens, D. H., Codomo, C. A. & Henikoff, S. Multifactorial profiling of epigenetic landscapes at single-cell resolution using MulTI-Tag. Nat. Biotechnol. 41, 708–716 (2023).
Stuart, T. et al. Nanobody-tethered transposition enables multifactorial chromatin profiling at single-cell resolution. Nat. Biotechnol. 41, 806–812 (2023).
Bartosovic, M., Kabbe, M. & Castelo-Branco, G. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol. 39, 825–835 (2021).
Xiong, H., Wang, Q., Li, C. C. & He, A. Single-cell joint profiling of multiple epigenetic proteins and gene transcription. Sci. Adv. 10, eadi3664 (2024).
Vangala, P. et al. High-resolution mapping of multiway enhancer–promoter interactions regulating pathogen detection. Mol. Cell 80, 359–373 (2020).
Arrastia, M. V. et al. Single-cell measurement of higher-order 3D genome organization with scSPRITE. Nat. Biotechnol. 40, 64–73 (2022).
Goronzy, I. N. et al. Simultaneous mapping of 3D structure and nascent RNAs argues against nuclear compartments that preclude transcription. Cell Rep. 41, 111730 (2022).
Donnard, E. et al. Comparative analysis of immune cells reveals a conserved regulatory lexicon. Cell Syst. 6, 381–394 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Robinson, J. T. et al. Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26 (2011).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Schmidl, C., Rendeiro, A. F., Sheffield, N. C. & Bock, C. ChIPmentation: fast, robust, low-input ChIP–seq for histones and transcription factors. Nat. Methods 12, 963–965 (2015).
Daley, T. & Smith, A. D. Predicting the molecular complexity of sequencing libraries. Nat. Methods 10, 325–327 (2013).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Hammal, F., Langen, P., de Bergon, A., Lopez, F. & Ballester, B. ReMap 2022: a database of human, mouse, Drosophila and Arabidopsis regulatory regions from an integrative analysis of DNA-binding sequencing experiments. Nucleic Acids Res. 50, D316–D325 (2021).
McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw. 3, 861 (2018).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Dreos, R., Ambrosini, G., Groux, R., Cavin Périer, R. & Bucher, P. The Eukaryotic Promoter Database in its 30th year: focus on non-vertebrate organisms. Nucleic Acids Res. 45, D51–D55 (2017).
Frankish, A. et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019).
Satopää, V., Albrecht, J., Irwin, D. & Raghavan, B. Finding a ‘kneedle’ in a haystack: detecting knee points in system behavior. In 2011 31st International Conference on Distributed Computing Systems Workshops 166–171 (IEEE, 2011).
Liang, K., Patil, A. & Nakai, K. Discovery of intermediary genes between pathways using sparse regression. PLoS ONE 10, e0137222 (2015).
Tommaso, P. D. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
Kluger, Y., Basri, R., Chang, J. T. & Gerstein, M. Spectral biclustering of microarray data: coclustering genes and conditions. Genome Res. 13, 703–716 (2003).
Virtanen, P. et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272 (2020).
Zitnik, M. & Zupan, B. NIMFA: a Python library for nonnegative matrix factorization. J. Mach. Learn. Res. 13, 849–853 (2012).
Zheng, R. et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 47, D729–D735 (2019).
Yeh, B. & Goronzy, I. GuttmanLab/chipdip-pipeline: Nature Genetics (2024) paper release (v1.0_publication). Zenodo https://doi.org/10.5281/zenodo.13952458 (2024).
Acknowledgements
We thank S. Hiley for editing. We thank I.-M. Strazhnik and A. Koivula for illustrations and formatting the figures. This work was funded by grants from the NIH (R01 HG012216, R01 DA053178, U01 DK127420 to M.G.), the Chan Zuckerberg Initiative Ben Barres Early Career Acceleration Award, the NIH UCLA-Caltech Medical Scientist Training Program (T32GM008042, I.N.G. and B.T.Y.), NCI F30CA278005 (J.K.G.) and the University of Southern California MD/PhD program (J.K.G.). Sequencing was performed at the Millard and Muriel Jacobs Genetics and Genomics facility at Caltech with support from I. Antoshechkin and at the Broad Institute Genomics Platform.
Author information
Authors and Affiliations
Contributions
A.A.P., M.R.B. and M.G. conceived ChIP-DIP; A.A.P. and M.R.B. developed ChIP-DIP; A.A.P., I.N.G. and J.K.G. optimized ChIP-DIP; A.A.P. and I.N.G. generated the data presented in this paper; C.S.L., O.E. and A.B. cultured, collected and treated cells; I.N.G. developed the computational pipeline; B.T.Y. generated the GitHub repository for the pipeline; I.N.G. performed data analysis and visualization; A.A.P., I.N.G. and M.G. generated figures and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
M.G., A.A.P., M.R.B., I.N.G. and J.K.G. are inventors of a submitted patent covering the ChIP-DIP method. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Potential sources of mixing in ChIP-DIP.
(a) Schematic of labeling strategy to generate Protein G beads coupled with a unique antibody-identifying oligonucleotide and a matched antibody. (i) Protein G beads are covalently modified with a biotin, (ii) oligonucleotides containing a 3’ biotin are conjugated to streptavidin, (iii) oligo-streptavidin complexes are mixed with biotinylated protein G beads and (iv) protein G beads are mixed with antibodies. This process is repeated for each unique oligonucleotide-antibody pair and then all bead-antibody conjugates are pooled together. (b) Schematic of three potential sources of dissociation of chromatin-antibody-bead-oligo conjugates that could lead to mixing during ChIP-DIP: dissociation 1) between oligo and bead, 2) between antibody and bead, or 3) between antibody and chromatin. (c) If oligos dissociate from their original beads and bind to distinct beads (oligo-bead dissociation), we would expect multiple distinct oligo types on the same bead. To quantify this, we computed the percent uniqueness of oligo-types within each split-pool cluster. The cumulative distribution of the uniqueness of antibody-ID oligos type (x-axis) within individual clusters is shown. (d) If antibodies dissociate from their original bead and reassociate with a different bead (antibody-bead dissociation), we expect that chromatin would associate with empty beads present in the experiment. We show a schematic of the experimental design to test for antibody movement between beads (top) and the quantification of reads per bead assigned to true targets (CTCF) or empty beads added during experimental processing steps (bottom). (e) If proteins (and their crosslinked chromatin) dissociate and reassociate to other beads containing the same epitope-specific antibodies (antibody-chromatin dissociation), we would expect that chromatin purified independently from human and mouse lysates would mix during the procedure. We show a schematic of the human-mouse mixing experimental design to test for chromatin movement (left) and quantification of species-specific reads assigned to human or mouse beads (right).
Extended Data Fig. 2 Mapping multiple components of the same regulator complex within a single experiment.
(a) Visualization of various components of the PRC1 (RING1B, CBX8) and PRC2 (EZH2, SUZ12, EED) complexes that were mapped within the same ChIP-DIP pool (K562 52 Antibody Pool) along a genomic region (hg38, chr4:500,000-5,500,000).
Extended Data Fig. 3 Histone modifications associated with five chromatin states.
(a) UMAP embedding of 12 histone modifications measured in K562 correspond to five chromatin states. (b) Metaplot of signal distribution of H3K36me3, H3K79me1 and H3K79me2 across the gene body of protein coding genes in K562. (c) Correlation scatterplot of H3K9Ac and H3K4me3 signals at promoter sites in mESC. (d) Enrichment heatmap of H3K9me3 and H4K20me3 at various associated (ZNF genes, LTRs, LINES) and unassociated (SINES, TSS) genomic elements in K562. H3 is shown as reference. For A-D, see Methods for details on ChIP-DIP experiments used for each analysis.
Extended Data Fig. 4 Chromatin regulators co-localizing with known histone targets.
(a) Metaplots of read coverage for three H3K4me3-associated chromatin regulators (JARID1A, RBBP5, PHF8) and H3K4me3 at four promoter groups in mESC. Promoter groups were identified using k-means clustering of CR signal. (b) Metaplot showing colocalization of multiple PRC1 and PRC2 members and their respective histone modifications at RING1B sites in K562. (c) Genome-wide correlation matrix of multiple HP1 proteins versus heterochromatin and euchromatin markers in K562. For A-C, see Methods for details on ChIP-DIP experiments used for each analysis.
Extended Data Fig. 5 Simultaneous mapping of distinct RNA polymerases and their isoforms.
(a) Bar graph showing enrichment of gene class coverage (rRNA, mRNA, snRNA or tRNA) for RNAP I, II and III in mESC. For each RNAP, the bar of its associated class (or classes) is highlighted. (b) Visualization of RNAP II phosphorylation isoforms across the NUP214 gene in K562 (left). Metaplot of signal distribution of RNAP II phosphorylation isoforms across the gene body of protein coding genes in K562 (right).
Extended Data Fig. 6 Chromatin dynamics and the relationship to gene expression following LPS stimulation in mDCs.
(a) Heatmap of change in normalized coverage per 100 kb bin for various mapped factors. For each factor, only enriched bins are shown and bins are sorted left-to-right by magnitude of change. (b) Violin plot of gene expression fold change for 6hrs vs 0hrs (left) and 24hrs vs 0hrs (right) grouped by sets of genes corresponding to sets of regions from Fig. 5C(see Methods). Shown are Mann-Whitney U test p-values. (c) Track visualization of H3K27ac at 0hrs, 6hrs and 24hrs across a genomic region (mm10, chr5:29,838,000-30,024,000) upstream of the inflammatory gene IL6 and containing regions belonging to the ‘activated’ set from Fig. 5B. (d) Heatmap of spearman correlation coefficients between histone coverage change and gene expression change between time points. Change is defined as the ratio between the two time points. All genes were included in the correlation heatmap on the left; only genes with a fold change of >2 in gene expression were included in the correlation heatmap on the right (see Methods and Supplemental Methods).
Extended Data Fig. 7 Transcription levels of specific clusters of H3K4me3 enriched regions.
(a) Violin plot of the transcriptional levels, measured by the RNAP II occupancy, of the five major clusters of H3K4me3 regions identified in Fig. 7.
Extended Data Fig. 8 Histone acetylation marks are highly correlated genome-wide.
(a) Genome-wide pearson correlation coefficients of 15 different histone acetylation marks in mESC. Correlations are based on coverage computed in 10 kb windows. (b) Comparison of 15 different histone acetylation marks across a genomic region (mm10, chr1:55,048,000-55,148,000) in mESC.
Extended Data Fig. 9 Enrichment profiles for NMF generated combinations (C1-C5) of histone acetylation marks.
(a) RNAP II, TF and CR enrichment matrix for regions assigned to combinations (C1-C5) from NMF decomposition of highly acetylated regions using histone acetylation marks, shown in Fig. 8. (b) Heatmap of genome position enrichments relative to TSS for regions assigned to combinations. (c) Transcription factors of top 10 most significant sequence motifs for regions assigned to each combination are listed.
Extended Data Fig. 10 Profiles for high density regions of NANOG-OCT4-SOX2.
(a) Plot showing normalized region scores (x-axis) for peak regions of NANOG-OCT4-SOX2, ordered by rank (y-axis). High density regions are defined as regions past the point where the slope = 1. (b) Track visualization of NANOG-OCT4-SOX2 upstream of the gene for the pluripotency transcription factor KLF4 in mESC. A high density region is indicated with a red bar; low density regions are indicated with grey bars. (c) Visualization of NANOG-OCT4-SOX2 near the TET2 gene, a developmentally associated chromatin regulator, in mESC. A high density region internal to the gene is indicated with a red bar. (d) Coverage metaplots over low density regions (LDR) vs high density regions (HDR) for pluripotency transcription factors and other transcriptional-related factors. Metagenes are centered on the region and the lengths represent the approximate difference in mean lengths (500 bps for LDRs and 14,500 bps for HDRs). An additional 4 kb surrounding each region is shown. (e) Enrichment heatmap for GO terms of genes associated with HDRs or LDRs containing C4, C5 or neither C4/C5 chromatin signatures. (f) Enrichment heatmap for development-associated GO terms of genes associated with HDRs or LDRs containing C4, C5 or neither C4/C5 chromatin signatures.
Supplementary information
Supplementary Information
Supplementary Figs. 1–13, Notes 1–3, Methods and Tables 1–5.
Supplementary Data 1
Antibody ID oligonucleotide sequences.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Perez, A.A., Goronzy, I.N., Blanco, M.R. et al. ChIP-DIP maps binding of hundreds of proteins to DNA simultaneously and identifies diverse gene regulatory elements. Nat Genet 56, 2827–2841 (2024). https://doi.org/10.1038/s41588-024-02000-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-024-02000-5
This article is cited by
-
Unravelling the complexity of gene regulation through multiplexed protein mapping
Nature Reviews Molecular Cell Biology (2025)
-
Split-pool barcoding serves up an epigenomic smorgasbord
Nature Genetics (2024)
