
Abstract
We conducted a multiancestry genome-wide association study of prostate-specific antigen (PSA) levels in 296,754 men (211,342 European ancestry, 58,236 African ancestry, 23,546 Hispanic/Latino and 3,630 Asian ancestry; 96.5% of participants were from the Million Veteran Program). We identified 318 independent genome-wide significant (P ≤ 5 × 10−8) variants, 184 of which were novel. Most demonstrated evidence of replication in an independent cohort (n = 95,768). Meta-analyzing discovery and replication (n = 392,522) identified 447 variants, of which a further 111 were novel. Out-of-sample variance in PSA explained by our genome-wide polygenic risk scores ranged from 11.6% to 16.6% for European ancestry, 5.5% to 9.5% for African ancestry, 13.5% to 18.2% for Hispanic/Latino and 8.6% to 15.3% for Asian ancestry and decreased with increasing age. Midlife genetically adjusted PSA levels were more strongly associated with overall and aggressive prostate cancer than unadjusted PSA levels. Our study highlights how including proportionally more participants from underrepresented populations improves genetic prediction of PSA levels, offering potential to personalize prostate cancer screening.
Similar content being viewed by others


Main
Prostate-specific antigen (PSA) is a KLK3-encoded prostate gland-secreted protein1,2,3 often elevated in those with prostate cancer. However, elevated levels can also be caused by other factors, such as benign prostatic hyperplasia (BPH), local inflammation or infection, prostate volume, age and germline genetics2,4,5,6,7. PSA screening for prostate cancer was approved by the Food and Drug Administration in 1994, but it is unclear if the benefits for prostate cancer-specific mortality reduction outweigh the harms from overdiagnosis and treatment of clinically inconsequential disease8,9,10,11. An estimated 20% to 60% of screen-detected prostate cancers are overdiagnoses (that is, prostate cancer that would not otherwise clinically manifest or result in prostate cancer-related death)12; an estimated 229 individuals must be invited to screen and 9 diagnosed to prevent 1 death13. The United States14, Canada15 and the United Kingdom16 recommend against universal population-based screening. Adjusting PSA for individuals’ predispositions in the absence of prostate cancer could improve the specificity (to reduce overdiagnosis) and sensitivity (to prevent more deaths) of screening.
Twin studies estimate PSA heritability to be 40% to 45%17,18, and genome-wide evaluations estimate heritability to be 25% to 30%19, suggesting that incorporating genetic factors may improve screening. Our recent work based on 85,824 European ancestry (EUR) and 9,944 non-EUR men found that genetically adjusted PSA (that is, inflated/deflated due to an individual’s genetic variants) most improved PSA screening discrimination for aggressive tumors19. We also identified 128 genome-wide significant (P < 5 × 10−8) variants explaining up to 7% of PSA variation in EUR, suggesting that many more PSA loci remain. Genome-wide polygenic risk scores (PRSs) explained up to 10% in EUR; however, the PRSs were less predictive in other groups, especially African ancestry (AFR; 1–3%). Additional variant discovery with larger, more diverse cohorts could provide novel insights into PSA genetic architecture and further improve prostate cancer screening.
Results
Composition of discovery and replication cohorts
Our discovery cohort consisted of 296,754 prostate cancer-free men from 9 cohorts not previously included in PSA GWAS, with 211,342 EUR (71.2%), 58,236 AFR (19.6%), 23,546 Hispanic/Latino (HIS/LAT; 7.9%) and 3,630 Asian ancestry (ASN; 1.2%); the Million Veteran Program (MVP) comprised 96.5%. Analytic workflow, genotype platforms, demographics and quality control data are available (Fig. 1 and Supplementary Tables 1–3). The pooled mean age at PSA measurement was 57.4 years (standard deviation (s.d.) = 9.6 years), and median PSA was 0.84 ng ml−1. For replication, we used previous results from 95,768 independent individuals19, including 85,824 EUR, 3,509 AFR, 3,098 HIS/LAT and 3,337 ASN (Supplementary Table 3).

The discovery GWAS analysis revealed 318 genome-wide significant (P < 5 × 10−8, two-sided test) variants associated with PSA, of which 184 were novel. The joint analysis (consisting of the discovery and replication cohorts) revealed 447 genome-wide significant variants associated with PSA, of which an additional 111 were novel. Both discovery and joint GWAS results were used to develop PRSs for PSA, which were then evaluated in GERA (when out of sample), PCPT and SELECT. Sig, significant; SNP, single-nucleotide polymorphism.
Discovery GWAS analysis of PSA-associated variants
In our discovery, we identified 318 independent genome-wide significant variants (264 EUR, 51 AFR, 17 HIS/LAT and 2 ASN) in a multiancestry analysis of ln-transformed PSA (Fig. 2, Supplementary Fig. 1 and Supplementary Tables 4 and 5) using multiple reference panels to account for different ancestries (Methods). Among them, 184 independent variants selected by mJAM20 were novel (to the best of our knowledge; Methods). Of the novel variants, 57 replicated at a Bonferroni level (P < 0.05/184 = 0.00027, same effect direction), an additional 80 replicated at P < 0.05 (and same direction), 43 demonstrated the same effect direction (but P > 0.05) and 4 showed no indication of replication (opposite effect direction). On average, compared to nonreplicated variants, replicated variants had slightly larger effect sizes (mean β = 0.30 versus 0.27) and were slightly more precise (mean standard error = 0.0039 versus 0.0042).

Concentric tracks are colored based on results from individual ancestries, with gray indicating results from the overall discovery meta-analysis (n = 296,754). The 100,000 variants with the smallest P values per ancestry are shown as points; larger circled points indicate the 318 genome-wide significant variants (P < 5 × 10−8; 184 of which were novel) from the overall discovery analysis across ancestries. Variant density in 10 Mbp bins from the overall analysis is shown as a heatmap above the overall track. The outermost ring displays genes associated with novel discovery PSA variants. Results are from a fixed effects meta-analysis of linear regression analysis with two-sided tests.
Of the 184 variants novel in the multiancestry discovery analysis, 112 were genome-wide significant in EUR, 8 in AFR and none in ASN or HIS/LAT (likely due to low sample size; Extended Data Fig. 1). Of the 8 in AFR, only 2 were frequent enough (Methods) to be assessed in other ancestry groups: rs2071041 (ITIH4; βAFR = 0.0237, 95% confidence interval (CI) = 0.0152–0.0322, pAFR = 4.9 × 10−8) that was also genome-wide significant in EUR (βEUR = 0.0180, 95% CI = 0.0124–0.0235, pEUR = 2.6 × 10−10, minor allele frequency (MAF) = 23.7%) and rs1203888 (LINC00261; βAFR = −0.0423, 95% CI = −0.0539 to −0.0307, pAFR = 8.7 × 10−13) that was not significant in EUR (pEUR > .05, MAF = 0.8%). The latter variant showed similar effect magnitude but was not Bonferroni significant in discovery HIS/LAT (βHIS/LAT = −0.0748, 95% CI = −0.120 to −0.0297, pHIS/LAT = 0.0012, MAF = 3.1%) and was not significant in discovery ASN (P > 0.05, MAF = 3.5%) or the replication cohorts (P > 0.05) (Supplementary Table 4). The remaining 6 AFR variants were too rare to be assessed in EUR. The variant rs184476359 (AR, multiancestry discovery β = −0.0590, 95% CI = −0.0774 to −0.0406, P = 3.4 × 10−10; replication β = −0.0870, 95% CI = −0.1370 to −0.0371, P = 6.3 × 10−4) was common in AFR (MAF = 17.7%), less common in HIS/LAT (MAF = 1.1%) and not adequately polymorphic to be imputed in ASN. Three variants in genes encoding PSA (rs76151346, βAfrican = 0.0821, 95% CI = 0.0577–0.107, pAfrican = 4.6 × 10−11, KLK3; rs145428838, βAFR = 0.224, 95% CI = 0.165–0.284, pAFR = 1.4 × 10−13, KLK3; rs182464120 βAFR = −0.213, 95% CI = −0.278 to −0.147, pAFR = 2.0 × 10−10, KLK2) exclusively imputed in AFR (all MAF < 5%, two <1%) did not exhibit strong evidence of replication in AFR (P > 0.05). The remaining two variants identified in AFR (rs7125654, βAFR = −0.384, 95% CI = −0.0489 to −0.0279, pAFR = 7.0 × 10−13; rs4542679, βAFR = 0.0422, 95% CI = 0.0288–0.0557, pAFR = 7.9 × 10−10) were more common (MAF > 5%) but also did not replicate (P > 0.05). Further, rs7125654 (TRPC6) was less common in HIS/LAT, but more common in ASN, and rs4542679 (RP11-345M22.3) was less common in HIS/LAT and not adequately polymorphic in ASN.
We next tested for effect size differences across ancestry groups for the 184 novel variants. Only rs12700027 (BRAT1/LFNG, I2 = 84.8%, P = 0.00019) demonstrated Bonferroni-significant heterogeneity (P < 0.05/184 = 0.00027). The variant had a strong EUR discovery effect (βEUR = 0.0327, 95% CI = 0.0247–0.0407, pEUR = 1.2 × 10−15, MAF = 0.10) but was not significant in other groups (βAFR = 0.0131, 95% CI = −0.0190 to 0.0452, pAFR = 0.42, MAF = 0.021; βASN = −0.176, 95% CI = −0.333 to 0.0203, pASN = 0.027, MAF = 0.021; βHIS/LAT = −0.0102, 95% CI = −0.0326 to 0.0121, pHIS/LAT = 0.37, MAF = 0.120). In our replication, the variant nominally (that is, P < 0.05) replicated (P = 0.0065, β = 0.0175, 95% CI = 0.0049–0.0302; pEUR = 0.003, β = 0.0327, 95% CI = 0.0247–0.0407) and showed no statistically significant evidence of differences across ancestry groups (I2 = 0.0%, P = 0.44), although sample sizes for detecting differences were smaller.
In silico assessment of potential functional features revealed that 20 of the novel variants (10.8%) were prostate tissue expression quantitative trait loci (eQTLs) and another 65 (35.3%) were eQTLs in other tissues (Supplementary Table 4). Five novel variants were missense and predicted deleterious, with Combined Annotation Dependent Depletion (CADD) scores >20 (Supplementary Table 4): rs11556924 in ZC3HC1, which regulates cell division onset; rs74920406 in ELAPOR1, a transmembrane protein; rs2229774 in RARG, in the hormone receptor family; rs113993960 (delta508) in CFTR, a causal mutation for cystic fibrosis21; and rs2991716 upstream of LOC101927871. An additional 11 variants were predicted to have high pathogenicity (CADD scores >15; Supplementary Table 4).
Replication of previously reported variants in discovery
When we tested 128 previously identified variants19 in our discovery cohort, 106 (82.8%) replicated with genome-wide significance, an additional 15 (11.7%) Bonferroni (P < 0.05/128 = 0.00039), an additional 6 at P < 0.05 (4.7%) and 1 variant flipped effect direction (Supplementary Table 6). Replication was highest for EUR, likely due to sample size, with 94 variants (73%) reaching genome-wide significance, an additional 22 variants (17.2%) meeting a Bonferroni-corrected level and 8 (6.3%) additional variants meeting P < 0.05 (Supplementary Table 6). Replication rates within AFR, our next largest group, were lower: 16 (12.5%) were genome-wide significant, 26 others (20.3%) met Bonferroni, an additional 39 (30.5%) had P < 0.05, 32 additional (25.0%) were in the same direction and 15 (11.7%) were in the opposite direction. Estimated rates were similar for HIS/LAT and lowest for ASN. Lastly, 16 of the 128 known variants showed heterogeneity across the four groups (Bonferroni-corrected P < 0.05/128 = 0.00039).
Joint meta-analysis of discovery and replication cohorts
In the multiancestry analysis including the discovery and replication cohorts, we identified 447 independent variants (409 EUR, 56 AFR, 22 HIS/LAT and 6 ASN, including 46 in >1 group; Fig. 3, Supplementary Fig. 1 and Supplementary Tables 7 and 8). Among the 111 variants that were novel (to the best of our knowledge) even relative to discovery alone, none showed evidence of ancestry effect size differences (P > 0.05/111 = 0.00045). Fifty-six (50.4%) of the 111 were genome-wide significant in EUR, but none were genome-wide significant in a non-EUR group (Supplementary Table 8). Allele frequencies and effect sizes of the novel variants largely followed those expected by power curves (Fig. 4).

Only genome-wide significant associations (P < 5 × 10−8) are plotted. The joint analysis (n = 296,754 discovery, plus n = 95,768 replication) detected 447 independent genome-wide significant PSA-associated variants. These included 111 novel variants that were conditionally independent from previous findings and the discovery-only analyses (indicated by the circles). Gene labels are given for variants with CADD > 15 and/or variants that are prostate tissue eQTLs. Results are from a fixed effects meta-analysis of GWAS performed using linear regression. All P values are two-sided.

Each point represents one of the 447 independent genome-wide significant variants identified in our mJAM multiancestry GWAS joint meta-analysis (n = 392,522). The estimated variant effect sizes are expressed in ln(PSA) per minor allele. The curves indicate the hypothetical detectable variant effect sizes for a given MAF, assuming statistical power of 80% and α = 5 × 10−8 (genome-wide significant), and assuming that the sample size of each of our populations is as follows: 297,166 EUR, 61,745 AFR, 6,967 ASN and 26,644 HIS/LAT. Effects are from a meta-analysis of GWAS performed using linear regression.
In the joint meta-analysis, 12 (10.8%) novel variants were prostate tissue eQTLs, and 50 (45.0%) additional variants were eQTLs for other tissues. Two were missense substitutions (Supplementary Table 7): rs1049742 in AOC1 and rs74543584 in MPZL2. Three additional novel variants had CADD scores>15: rs1978060, an eQTL for TBX1 in prostate tissue; rs339331 an eQTL for FAM162B in adipose tissue; and rs57580158, an intergenic variant with evidence of conservation.
Medication sensitivity analysis
A sensitivity analysis in the UK Biobank (UKB) excluded individuals taking medications that could affect PSA (that is, 5-alpha reductase inhibitors and testosterone). For PSA-associated variants, our primary results in the UKB were highly correlated (R = 0.93, Extended Data Fig. 2) with the sensitivity analyses, suggesting these medications did not impact our results.
Out-of-sample PSA variance explained by PRSs
We evaluated different strategies for constructing PRSs for PSA first using discovery results (Methods). For testing these PRSs, four cohorts without prostate cancer were out-of-sample: Kaiser Permanente’s Genetic Epidemiology Research on Adult Health and Aging (GERA), the Selenium and Vitamin E Cancer Prevention Trial (SELECT)22, the Prostate Cancer Prevention Trial (PCPT)23 and All of Us (AOU)24.
In GERA, PRS318, constructed from the 318 independent genome-wide significant variants in the multiancestry meta-analysis, generally had higher variance explained when using longitudinal measurements, rather than earliest PSA, with 13.9% (95% CI = 13.1%–14.6%) in EUR (n = 35,322), 13.1% (95% CI = 10.6%–15.6%) in HIS/LAT (n = 2,716), 9.3% (95% CI = 6.8%–12.0%) in AFR (n = 1,585) and 9.0% (95% CI = 7.0%–11.4%) in ASN (n = 2,518). The variance explained in the other three cohorts was ~3–6% lower depending on the group (Supplementary Table 9).
Expanding to a genome-wide approach, PRS-CSx (PRSCSx-disc; included more than genome-wide significant variants; 1,070,230 variants; Methods) resulted in improved predictive performance. The variance explained increased to 16.6% (95% CI = 15.9%–17.5%) in EUR and 18.2% (95% CI = 15.4%–20.8%) in HIS/LAT (Fig. 5a and Supplementary Table 9). The relative increase was largest in ASN, with variance explained reaching 15.3% (95% CI = 12.7%–18.1%), and smallest in AFR, with variance explained 8.5% (95% CI = 6.1%–11.0%).

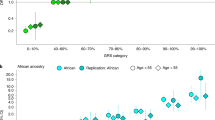
PRSs for PSA trained on the discovery GWAS meta-analysis was evaluated in GERA, in addition to the three main validation cohorts (PCPT, SELECT and AOU). Joint PRSs were trained on summary statistics from the meta-analysis of the discovery GWAS and replication GWAS. A PRS was first constructed based on independent variants identified using mJAM that reached genome-wide significance. Then a multiancestry genome-wide score was developed using PRS-CSx. a,b, The variance explained by genome-wide PRSs was up to 16.9% in EUR, 18.6% in HIS/LAT, 9.5% in AFR and 15.3% in ASN (a) and decreased as age increased (b). Estimates and full details are found in Supplementary Table 9. Error bars indicate 95% CIs. Variance explained was estimated from partial r2 estimates from linear regression models adjusted for age and genetic ancestry PCs. Sample sizes of each population are specified in a. EUR, European; LAT, Hispanic/Latino; AFR, African; EAS, East Asian; ASN, Asian; OTH, other; AMR, Admixed American; Disc., discovery.
Second, we developed PRSs for PSA using the results from the joint GWAS meta-analysis (n = 392,522), which combined the discovery meta-analysis with previously published results from Kachuri et al.19. These scores were validated in PCPT, SELECT and AOU, but not GERA, which was included in the previously published meta-analysis and therefore not out of sample.
For the independent genome-wide significant PRSs, PRS318 explained 9.5% (8.8%–10.3%) of variation in baseline PSA in SELECT EUR (n = 22,173), whereas PRS447 (from the 447 independent genome-wide significant variants identified in the joint meta-analysis) explained 10.9% (10.2%–11.8%), which exceeded the 8.5% (95% CI = 7.8%–9.2%) of variance explained by PRS128 (from the 128 independent variants described in our prior GWAS of 95,768 men19). Variance explained in PCPT EUR (n = 5,725) was slightly lower. In AOU EUR (n = 11,922), variance explained was slightly higher, with PRS128 explaining 8.6% (95% CI = 7.7%–9.6%), PRS318 explaining 9.6% (95% CI = 8.6%–10.6%) and PRS447 explaining 11.3% (95% CI 10.2%–12.4%).
Removing individuals with BPH (known to influence PSA) did not appreciably change differences across cohorts; however, variance explained was slightly higher in all populations (<0.5% higher), albeit with overlapping CIs (Supplementary Table 9).
Among SELECT AFR (n = 1,173), PRS128 explained 3.4% (95% CI = 1.6%–5.8%), PRS318 explained 6.5% (95% CI = 4.0%–9.5%) and PRS447 explained 7.0% (95% CI = 4.5%–10.1%) of variance; PRS447 more than doubled variance explained by PRS128. AOU AFR (n = 2,471) estimates were 1–2% smaller.
A genome-wide PRS-CSx (PRSCSx-joint) compared to PRSCSx-disc modestly increased variance explained by ~1–1.5% in EUR in PCPT (11.6%, 95% CI = 10.0%–13.1%), SELECT (13.9%, 95% CI = 13.1%–14.9%) and AOU (14.7%, 95% CI = 13.5%–16.0%). PRSCSx-joint also improved ~3% upon the PRS previously reported by Kachuri et al.19, estimated here to be 8.6% in PCPT and 10.4% in SELECT for PRS-CSx (PRSCSx-Kachuri, Supplementary Table 9). Among SELECT AFR, PRSCSx-joint showed no improvement (7.2%, 95% CI = 4.6%–10.0%) over PRSCSx-disc, whereas variance explained in AOU increased by 0.3% (5.8%, 95% CI = 4.1%–7.8%). Notably, PRSCSx-joint yielded a substantial improvement upon the previously published PRSCSx-Kachuri estimates19 of 1.64% in SELECT, although this was still under half of that observed in EUR.
In SELECT EUR, PRS-CSx explained 13.9% of variation, whereas PRS447 explained 10.9% of the variation. Assuming that variance explained is nested between these approaches, we estimate 78.4% (10.9%/13.9%) of PRS-CSx variation may be explained by PRS447. This is expected since information across the different PRSs overlaps, and the initial genome-wide significant variants from our large-scale GWAS are the most informative for explaining variation in PSA.
Third, we examined how the PSA PRS variance explained varied by age. These analyses were performed in GERA to have a large enough sample size in each age group and used PRSCSx-disc to provide out-of-sample estimates. The estimated variance explained by the PRSs decreased with increasing age in all GERA ancestry groups, albeit with somewhat wide CIs (Fig. 5b and Supplementary Table 10); for example, PRSCSx-disc explained 16.4% (95% CI 14.6%–18.5%) of variation in PSA among EUR < 50 years, and this decreased to 8.7% (95% CI 7.0%–10.5%) for men ≥80 years.
Finally, for PRSs constructed from genome-wide significant independent variants, variance explained using weights corresponding to effect sizes from the multiancestry meta-analysis was almost always equal to or higher than variance explained using ancestry-specific weights (Supplementary Table 10). This was observed for both the discovery (PRS318) and joint meta-analysis (PRS447). The few instances where the variance explained was estimated lower almost always had <1% difference and wide CIs around the estimate (that is, smallest sample sizes likely had unstable estimates).
Relationship of PSA PRSs with prostate cancer aggressiveness
In GERA, we performed a case-only analysis to examine the association between PSA PRSCSx,disc (the out-of-sample PRSs with the highest variance explained) and Gleason score. Results were consistent with previous work suggesting screening bias decreases the likelihood of identifying high-grade disease, whereby men with higher PRS values (indicating a genetic predisposition to higher constitutive PSA) are more likely to be biopsied, but less likely to have high-grade disease19; in EUR cases, a standard deviation increase in PRSCSx-disc was inversely associated with Gleason 7 (odds ratio (OR) = 0.78, 95% CI = 0.73–0.84, P = 1.2 × 10−13) and ≥8 (OR = 0.71, 95% CI = 0.64–0.79, P = 6.2 × 10−10) compared to Gleason ≤6. Other ancestry groups had similar estimated ORs, though not always statistically significant, likely owing to sample size (Supplementary Table 11; for example, AFR Gleason 7 (OR = 0.88, 95% CI = 0.67–1.17, P = 0.39) and ≥8 (OR = 0.65, 95% CI = 0.43–0.99, P = 0.043)).
Genetically adjusted PSA prostate biopsy eligibility impact
We examined how PRSCSx,disc would have changed biopsy recommendations for cases and controls, according to age-specific thresholds in GERA (Methods). In EUR individuals who had negative biopsy results (that is, controls, n = 2,378), 16.0% with unadjusted PSA levels exceeding age-specific thresholds for biopsy were reclassified to ineligible for biopsy. Among controls with PSA that did not indicate biopsy, 2.4% were reclassified to biopsy eligible, resulting in a control net reclassification improvement (NRI) of 13.6% (95% CI = 12.2%–15.0%; Fig. 6a and Supplementary Table 12). In individuals with positive biopsies (that is, cases; n = 2,358), 3.9% were reclassified to eligible, whereas 13.1% were reclassified to ineligible, resulting in a case NRI of −9.2% (95% CI = −10.3% to −8.0%). Of cases who became ineligible, 71.1% had Gleason scores ≤7, as compared to 56.5% who remained eligible (although we note some of these men may have had biopsies for reasons other than their PSA measurement (for example, abnormal digital rectal exam findings or strong family history)). In AFR controls (n = 110), 16.0% were reclassified to ineligible, whereas 2.4% were reclassified to eligible, resulting in an NRI of 3.6% (95% CI = 0.1% to 7.1%; Fig. 6b). In AFR cases (n = 310), 5.2% were reclassified to eligible and 6.8% were reclassified to ineligible, resulting in an NRI of −1.6% (95% CI = 3.0% to −0.2%). Other groups are also presented (Extended Data Fig. 3 and Supplementary Table 12). We obtained 8 years of additional follow-up on the 78 controls in all groups now classified as eligible; 3 men were later diagnosed with prostate cancer.

a,b, PSA values were adjusted (Methods) using the PRS-CSx estimate from the out-of-sample discovery cohort and assessed in GERA using age-specific cutoffs in European (n = 4,736; a) and African (n = 420; b). GERA Hispanic/Latino and Asian are shown in Extended Data Fig. 3. The Sankey diagram is based on percentages of each of the flows from/into nodes. N/A, not applicable.
To assess potential variability in genetic adjustment across PSA, we compared measured versus genetically adjusted PSA across a range of values in GERA (n = 43,945). We observed consistent relative adjustment on the ln scale (Extended Data Fig. 4); for example, at a measured PSA of 2.5, 6.5 and 10.0 ng ml−1, the genetically adjusted PSA interquartile range (IQR) ranged from 2.6 to 3.7, 4.6 to 6.8 and 9.1 to 12.5, respectively. These results suggest genetic adjustment is applicable at least to PSA values <20, although the implications are most profound around values where clinical decisions are made (for example, age-specific PSA thresholds).
Genetically adjusted PSA overall and aggressive prostate cancer impact
Previous work suggests midlife PSA predicts lethal prostate cancer25. In GERA EUR, genetically adjusted midlife ln PSA had a larger estimated association magnitude with overall prostate cancer (OR = 4.57, 95% CI = 4.27–4.88) than measured PSA (OR = 4.30, 95% CI = 4.04–4.58)), though the CIs overlapped. The difference was even larger for aggressive disease, with OR = 3.92 (95% CI = 3.54–4.35) for adjusted versus OR = 3.46 (95% CI = 3.15–3.81) for measured, though again CIs overlapped. AFR showed similar trends, with the genetically adjusted association with prostate cancer OR = 5.85 (95% CI = 4.73–7.23) versus measured OR = 4.72 (95% CI = 3.56–6.27), and the aggressive genetically adjusted OR = 5.39 (95% CI = 3.95–7.35) versus measured OR = 4.72 (3.56–6.27). Estimates in HIS/LAT were also similar, but ASN showed no difference (Supplementary Table 13). Cross-validated area under the curve estimates also showed essentially no difference between adjusted and measured PSA, with estimates ranging from 0.7 to 0.8 in the different groups (Supplementary Table 13).
Associations with previously reported prostate cancer variants
In our discovery cohort, 20 of our 184 novel PSA-associated variants (10.8%) were genome-wide significantly associated with prostate cancer in the PRACTICAL consortium’s EUR GWAS26 (Supplementary Tables 4 and 6), and 19 additional (10.3%) at a Bonferroni level (P < 0.05/184 = 0.00027). With bias correction related to more frequent screening in men with higher constitutive PSA (Methods)19,27, this count was reduced to 13 (7.0%) at genome-wide significance and an additional 14 (7.6%) at Bonferroni. Of the 111 novel PSA-associated variants from the meta-analysis, 8 (7.1%) were genome-wide significantly associated with prostate cancer, and an additional 11 (9.8%) at Bonferroni (P < 0.00045). With bias correction, 5 (4.5%) were genome-wide significant, and an additional 4 (3.6%) Bonferroni.
Associations with previously reported BPH variants
In discovery, one (rs1379553) of 137 variants was genome-wide significantly associated with BPH in a UKB EUR GWAS28. Eight additional met a Bonferroni level (P < 0.05/137 = 0.00036). Out of the 96 available joint meta-analysis-identified variants, 1 was genome-wide significant (rs627320) and 6 more met a Bonferroni level (P < 0.045/96 = 0.00052).
Associations with urinary symptom variants
In discovery-identified variants, rs12573077 (P = 8.4 × 10−5) met a Bonferroni level (P < 0.05/177 = 0.00028) for association with urinary symptoms in GERA (Supplementary Tables 4, 6 and 14). In the joint meta-analysis, none met a Bonferroni level (P< 0.05/110 = 0.00045).
PSA variant associations with prostate volume
Thirty-one of the 407 PSA variants tested demonstrated some evidence of an association with prostate volume in the Canary Prostate Active Surveillance Study (Canary PASS); rs182464120 was strongly associated (P = 2.0 × 10−11), rs12344353 met a Bonferroni level (P = 5.3 × 10−5 < 0.05/407 = 0.00012) and 29 other variants met a nominal level (P < 0.05) (Supplementary Table 15).
Associations with KLK3 plasma pQTL
Of the 447 variants from the joint meta-analysis, 409 had corresponding plasma protein quantitative trait loci (pQTL) association results for KLK3 from the UKB Pharma Proteomics Project29. In EUR (n = 46,214), GWAS and KLK3 pQTL effects were highly correlated (R = 0.85, P = 3.7 × 10−117) (Extended Data Fig. 5). Eleven variants were associated with relative KLK3 abundance at P < 0.05/409, and, as expected, the strongest two associations were in KLK3 (rs17632542, rs61752561) (Supplementary Table 16). Among AFR (n = 1,065), we observed an attenuated correlation with KLK3 abundance (R = 0.14, P = 0.0034), although no individual pQTL associations reached statistical significance.
Associations with eGenes
In our single-cell RNA sequencing analysis, eGenes for PSA-associated variants were expressed across prostate cells, especially in prostate luminal epithelial cells (produce PSA), as expected if the genes modify PSA (Supplementary Table 17). Extended Data Fig. 6 shows expression of the eQTL genes across multiple prostate tissue cell types, including luminal cells of the prostate epithelium and its precursor cells (for example, basal epithelial cells of prostate; expression sorted by KLK3). Percentile expression of eQTL-associated genes was significantly higher in luminal cells than all other prostate cell types (P = 0.0006), suggesting these genes are more active in this cell type than other prostate cells (Extended Data Fig. 7) and supporting the hypothesis that these eQTL genes are involved in PSA expression.
Discussion
Our PSA GWAS detected 448 genome-wide significant variants, including 295 that were novel (to the best of our knowledge, 184 in discovery and 111 in joint meta-analysis), nearly quadrupling the total number of associated variants. The variance explained by genome-wide PRSs ranged from 11.6% to 16.9% in EUR, 5.5%–9.5% in AFR, 13.5%–18.6% in HIS/LAT and 8.6%–15.3% in ASN. We also observed a decline in PRS predictive performance with increasing age, particularly the oldest ages. The majority of newly identified variants were uniquely associated with PSA and not prostate cancer.
Our discovery included more AFR individuals than any prior study of PSA genetics. Of the eight genome-wide significant variants identified in the discovery phase in AFR, only two were sufficiently common to be assessed in EUR; the rs1203888 (LINC00261) association was unique to AFR. These eight variants generally failed to meet replication Bonferroni significance, although the sample size was small (3,509 AFR); rs18447639 in the AR gene was closest to replicating. Androgen receptor (AR) signaling is required for normal prostate development and function, but is hijacked during carcinogenesis30. Because prostate tumor growth and progression depend on AR signaling, androgen deprivation therapy remains a frontline treatment for progressing prostate cancer, and AR activity inhibition may delay progression31.
Prostate tissue eQTLs were found at 10.9% of novel discovery and joint variants, and 49.7% were eQTLs in other tissues. In addition, 16 discovery variants and five meta-analysis variants predicted deleterious regulatory effects. Putative deleterious genes included: AOC1 (histamine metabolism regulator, non-steroidal anti-inflammatory drug sensitivity32,33), MPZL2 (thymus development, T cell maturation) and ZC3HC1 (cell cycle progression regulator, coronary artery disease susceptibility34,35). We also observed an association with the deltaF508 mutation in CFTR that causes cystic fibrosis, which is accompanied by infertility in 97% of affected males36 and has been linked to obstructive azoospermia (ClinVar37 accession SCV001860325). We detected another signal with possible links to male fertility, rs372203682 in LMTK2, a gene implicated in spermatogenesis38 that interacts with AR and inhibits its transcriptional activity39.
In SELECT, the PSA variance explained by our independently associated GWAS variants was ~1% larger than previously explained19 in EUR and ~3% higher in AFR. The variance explained in SELECT and PCPT was substantially less than that in GERA, even though we evaluated only variants from our discovery (which did not include GERA), likely due in part to selection criteria requiring PSA ≤ 3 ng ml−1 (SELECT)22 and ≤4 ng ml−1 (PCPT)23 at baseline. This was not required in AOU, yet variance explained for EUR was at most 0.5% higher than SELECT and thus also lower than GERA. For AOU AFR, variance explained was 2–3% lower than in SELECT, suggesting other factors may affect performance. Estimated variance explained was <0.5% higher when excluding men with a BPH diagnosis. By BPH, we mean a clinical diagnosis; most patients evaluated for potential prostate cancer have evidence of BPH, which can result in elevated PSA40,41. These findings highlight the need to evaluate genetically adjusted PSA in a wider range of clinical settings, as well as the challenges with curating out-of-sample cohorts with clinical data sufficient for such evaluations.
The performance of PRS constructed using weights from the multiancestry meta-analysis typically matched or surpassed that using ancestry-specific weights. As expected, genome-wide PRS-CSx generally achieved 1–6% higher variance explained than the PRSs limited to mJAM genome-wide significant variants. Improvement was not equal across populations and was largest in HIS/LAT, followed by EUR, ASN and then AFR. This difference may be due to several factors. First, PRS-CSx uses a single hyperparameter across ancestry groups, which may not capture different correlation structures. Second, HapMap3 variants used by PRS-CSx do not tag genetic variation equally well across ancestries. Fine-mapping PRS methods do not limit to this set of tagging variants and may be more likely to capture population-specific variants. Third, the choice of linkage disequilibrium (LD) reference panels has slightly different implications for the two approaches. PRS-CSx relies on LD reference panels for estimating joint variant effect sizes, whereas fine-mapping requires LD information for identifying independent variants from summary statistics. mJAM advances other fine-mapping approaches by incorporating population-specific LD to be more accurate than a single population20 or the largest ancestry group. Although PRS-CSx provides more flexibility to accommodate different genetic architectures, it may be more sensitive to LD reference panel choices and LD mismatches between training and testing populations, especially without a separate parameter tuning dataset.
Compared to previous work19, genetically adjusted PSA reduced unnecessary biopsies less, despite being in the same GERA population. Our previous study likely overestimated reclassification in controls because of partial train/test overlap (GERA was included); here we report results only without overlap. We also saw an increase in magnitude in genetically adjusted midlife PSA association with prostate cancer in most GERA groups, although CIs overlapped for all, and whereas our previous study19 did not see any benefit in AFR, we saw a numeric increase that was not statistically significant.
Our investigation had several limitations. Relative to prior PSA genetics studies, the discovery and replication cohorts included here substantially increased the number of men from diverse populations. Although both were very large (~300 K and ~100 K), the replication had disproportionately smaller AFR (discovery ~58 K, replication ~3.5 K) and HIS/LAT populations (~24 K and ~3 K). Nevertheless, for AFR, 43% of variants met a nominal replication threshold, many more than the 5% expected by chance. Going forward, our PSA Consortium will continue to seek new study populations with both genotypic and phenotypic data representing diverse participants. We also suspect that we had limited power to detect effect size heterogeneity, especially as variants that exhibited significant heterogeneity were mostly known variants in strongly associated regions. Another limitation is that GERA biopsy reclassification may have been specific to Kaiser Permanente clinical guidelines19. In addition, although we did our best to restrict relevant analyses to prostate cancer-free individuals, some likely had undetected prostate cancer42. However, the number was unlikely to be large enough to materially impact our results because our study population was relatively young; the average age among men in the MVP (comprising 96.5% discovery) was 58, 52, 54 and 54 years for EUR, AFR, HIS/LAT and ASN, respectively. Further, the PRSCSx PSA variance explained increased for younger ages. Most novel PSA-associated variants were not associated with prostate cancer, and those that were may have been due to screening bias19. The lack of BPH information in most of our cohorts was an additional limitation, but most novel variants associated with PSA were not associated with BPH in others’ work on UKB EUR28, and the variance explained by PRSs in SELECT was affected by <0.5% in participants with BPH. We were unable to account for prostate volume, a strong predictor of PSA43. Finally, we note that our GWAS and resulting PRSs were developed for total PSA. Future work should capture genetic factors specific to constituents of total PSA.
In summary, we undertook a multiancestry study with over three times the sample size of previous work19, expanding our understanding of the genetic basis of PSA and our potential to improve the accuracy of PSA genetic adjustment across ancestries. Using an ancestrally diverse population, we detected hundreds of novel variants associated with PSA that were largely independent of prostate cancer and BPH. These findings explain additional variation in PSA, especially among AFR men, who suffer the highest prostate cancer morbidity and mortality, as well as HIS/LAT men, which highlights the importance of studying diverse populations to enable novel discoveries and construct PRS that will perform equally across ancestry groups. Taken together, our work moved us closer to leveraging genetic information to personalize PSA and substantially improved our understanding of PSA across diverse ancestries.
Methods
Inclusion and ethics
The African American Prostate Consortium (AAPC) was approved by their institutional review board (IRB). The ethics review board of the Program for the Protection of Human Subjects of Mount Sinai School of Medicine approved the Mount Sinai BioMe Biobank (BioMe) (#HSD09-00030, #07-0529 0001 02 ME). The University of Chicago Biological Sciences Division IRB Committee A (#IRB12-1660) approved the Chicago Multiethnic Prevention and Surveillance Study (COMPASS). Local and national IRBs approved Men of African Descent and Carcinoma of the Prostate (MADCaP). The Multiethnic Cohort (MEC) was approved by their IRB. The VA Central IRB approved the MVP. The IRBs at Vanderbilt University and Meharry Medical College approved the Southern Community Cohort Study (SCCS). The Vanderbilt University Medical Center IRB approved BioVU. GERA was approved by the Kaiser Permanente Northern California IRB and the University of California, San Francisco. A local ethics committee approved the Malmö Diet and Cancer Study. The Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial was approved by the IRBs at each participating center and the National Cancer Institute, and the informed consent document allows data use for cancer and other adult disease investigations; we used publicly posted summary statistics, for which no IRB is required. The research was conducted with approved access to UKB data (#14105).
Written informed consent was obtained from all study participants. Participants received no compensation.
Discovery participants and phenotype measurements
Our primary analyses included 296,754 men from seven cohorts that had not previously been analyzed in studies of PSA genetics. These cohorts are described briefly below; additional details, including array, ancestry, imputation reference panels, sample sizes, number of variants and standard filters applied, are described in Supplementary Tables 1–3. To ensure participants had a functional prostate unaffected by surgery or radiation and to exclude individuals at a high risk of undiagnosed prostate cancer44, participants were restricted to men with no history of prostate cancer or surgical resections of the prostate and at least one PSA measurement between 0.01 and 10 ng ml−1. Analyses were based on each individual’s earliest recorded PSA level. For descriptive statistics, meta-analysis of PSA medians from each cohort was done with the weighted median of medians method in the R v4.2.3 (ref. 45) package metamediation v1.0.0 (ref. 46). Subpopulations were defined by self-identified race/ethnicity and/or genetically inferred ancestry, depending on the cohort.
The AAPC comprises AFR studies with prostate cancer phenotyping26. BioMe is a longitudinal cohort linked to Epic electronic health records (EHRs)47. Individuals were EUR, HIS/LAT or AFR. COMPASS is a longitudinal study of Chicagoans with >11,000 participants currently enrolled (82% African American)48 with PSA data49. MADCaP is a consortium of epidemiologic studies addressing the high prostate cancer burden in AFR men50,51. MEC is a prospective cohort study that enrolled >215,000 Hawaii/Los Angeles residents aged 45 to 75 years between 1993 and 1996 (refs. 52,53). MVP is a multiancestry cohort recruited nationwide. Information is obtained from EHRs, including inpatient International Classification of Diseases, Ninth Revision codes, Current Procedural Terminology (CPT) procedure codes, clinical laboratory measurements and reports of diagnostic imaging modalities54. Subpopulations were created using the harmonized ancestry and race/ethnicity method55. SCCS is a prospective cohort study that recruited 85,000 predominantly AFR adults from community health centers in the southeastern United States. This study included only men of AFR ancestry56.
Replication cohorts
Genome-wide significant variants identified in the discovery cohort were tested for replication in the previous largest GWAS of PSA, which included 95,768 men (85,824 EUR, 89.6%)19, using a Bonferroni-corrected α level. In addition, previously identified genome-wide significant variants19 were tested for replication in our independent discovery cohort. Statistical tests throughout were two-sided.
Additional PRS evaluation cohorts
For our discovery results, we evaluated PSA PRS performance and reclassification in individuals from GERA (also in the replication, out of sample for the discovery (n = 35,322; 28,503 EUR, 2,716 HIS/LAT, 2,518 ASN and 1,585 AFR)).
Additional out-of-sample cohorts for (both the discovery analysis and the joint meta-analysis of discovery and replication) PRS assessment was done in genotyped individuals from the PCPT23 (n = 5,725 EUR), SELECT22 (n = 25,366; 22,173 EUR, 1,763 AFR/EUR, 1,173 AFR and 257 ASN) and AOU (n = 17,512; 11,922 EUR, 2,469 AFR, 1,783 other and 1,336 HIS/LAT)24, which have been previously described. Briefly, PCPT and SELECT began as randomized, placebo-controlled, double-blinded clinical trials of finasteride and selenium and vitamin E, respectively, and both enrolled men ≥55 years. Individuals in SELECT and PCPT were required to have PSA ≤ 3 ng ml−1 (ref. 22) and ≤4 ng ml−1 (ref. 23), respectively, at baseline. The National Institutes of Health’s (NIH) AOU is committed to including groups that have been historically underrepresented in research24. From AOU, we selected individuals with PSA > 0.01 between the ages of 40 and 90 years, with short-read whole-genome sequencing (WGS) data and no survey or EHR conditions/observations reflecting a history of prostate cancer. The median PSA measurement we used was required to be ≤10 ng ml−1. PRSs were calculated with the WGS data restricted to variants with population-specific allele frequency ≥1% or a population-specific allele count >100 for any genetic ancestry. Genetic ancestry was determined using a random forest classifier trained on the principal component (PC) space of the Human Genome Diversity Project and 1000 Genomes Project (KGP)57.
Genotype quality control and imputation
Study participants were genotyped using conventional GWAS arrays (Supplementary Table 1). Genotypes were then imputed using imputation servers (Michigan imputation server v1.5.758, with Minimac4 v1.0.2 (ref. 59), Eagle v2.4 (ref. 60)), Minimac3 v2.0.1 (ref. 59) or IMPUTE2 v2.3.2 (ref. 61). The vast majority of studies imputed to the KGP phase 3 reference panel62, with one substudy imputing to KGP phase 1 just for the X chromosome63 and another imputing to the TOPMed r2 reference panel58. Because all but two studies (>95% of participants) used genome build 37, we lifted over the assembly of those from build 38 to build 37 using triple-liftOver64 v133 (2022-05-20), an extension of LiftOver65 that accounts for regions inverted between builds.
Standard genotype and individual-level quality control procedures were implemented in each ancestry group in each participating study. Specific study protocols are delineated in Supplementary Table 1, with additional quality control steps and details in Supplementary Table 2. Unless information was unavailable or a filter did not make sense for a particular group, variants were retained if their imputation quality score was ≥0.3, their MAF was ≥0.5% if the sample size was ≥1,000 and ≥5% otherwise, their Hardy-Weinberg equilibrium was ≥1 × 10−8, they were mapped in build 37 and they had an MAF difference ≤0.2 compared to KGP populations (full details in Supplementary Table 3). For the cohorts that meta-analyzed subcohorts (for example, the three small AFR sub-cohorts within the SCCS AFR group; Supplementary Table 2), we also required that variants be present in all sub-cohorts (necessary for multiancestry analysis method limitations, although this removed only a very small number of variants; Supplementary Table 3). Finally, we excluded variants if they were present in only one study with n < 2,000.
Association analyses
GWAS within each ancestry group in each study were undertaken using linear regression of ln PSA on additive genotypes and, when using multiple measurements, the long-term average residual by individual66. The minimum set of covariates included age at PSA measurement and genetic ancestry PCs. If available, GWAS also adjusted for batch/array, body mass index and smoking status (Supplementary Table 1). Meta-analyses of each ancestry group and across the overall discovery cohort were conducted using inverse-variance weighted fixed effects models using a custom-patched version of METAL v2011-03-25 that prevents numerical precision loss (lines 633 and 635 of ‘Main.cpp’ modified to the number 15 to output 15 digits precision)67. We also assessed heterogeneity with Cochran’s Q across the four ancestry groups.
To identify independently associated genome-wide significant (P ≤ 5 × 10−8) variants with computational efficiency, we first formed clumps of genome-wide significant variants such that all clumps were ≥10 Mb apart and independent of one another; specifically, the top variant was chosen, genome-wide significant variants ≤10 Mb from any variant in the clump were added to the clump, the process was iterated until a final clump was formed, and then the process was repeated to form more clumps (that is, clumps were created such that there was no additional genome-wide significant variant ≤10 Mb). Within each clump, we used mJAM v2022-08-05 (ref. 20), which uses population-specific LD reference panels for each contributing cohort and ancestry group to model the correlation among variants, with an r2 < 0.01 threshold in all ancestry groups. Genotypes using the appropriate GERA group (EUR, HIS/LAT, AFR and ASN) served as references68.
To maximize discovery efforts, we combined our discovery cohort (n = 296,754) with our replication cohort (n = 95,768), for a total of 392,522 individuals.
Associations were considered novel if they had low LD from all previously reported variants19. Specifically, we required r2 < 0.01 in all four ancestry groups, again using GERA as LD reference.
Annotation
Variants were annotated using FUMA v1.5.2 (ref. 69). We first prioritized genes that included a significant prostate eQTL from GTeX v8 (www.gtexportal.org). We then prioritized other significant eQTLs and finally by closest gene. Deleteriousness of mutations was determined by CADD scores; a recommended cutoff to identify potentially pathogenic variants of scores ≥15 has been suggested (the median of splice site changes and non-synonymous variants from CADD v1.0; corresponds to the top 3.2% of variants)70. Gene names follow canonical nomenclature in alignment with RefSeq v226 (ref. 71). Circos plots were generated using Circos v0.69-6 (ref. 72).
Medication sensitivity analysis
Some of our study participants may have taken medications that can affect PSA. In particular, 5-alpha reductase inhibitors and testosterone can impact PSA73,74. We assessed the use of these medications among 26,669 UKB EUR men with at least one PSA measurement. Men with a prescription for at least one of the two medications prior to PSA measurement were considered users. Ten percent of the men were prescribed 5-alpha reductase inhibitors and 0.56% testosterone. We also controlled for potential confounding by alpha blocker use.
Out-of-sample PRS variance explained
We calculated PRSs to assess the overall PSA variance explained by genetics, and to adjust PSA measurements for PSA genetics. All PRS results are shown only in independent cohorts (that is, training dataset completely independent of testing dataset), such that assessments of performance are unbiased. Nonparametric bootstrap percentile CIs for variance explained were calculated using 1,000 replicates.
We used two sets of individuals to construct the PRSs. First, we constructed PRSs from our discovery cohort to allow assessment in GERA, PCPT, SELECT and AOU. Second, we constructed PRSs from the meta-analysis of discovery and replication (which included GERA), with assessment in PCPT, SELECT and AOU only. For GERA, we included results using first and multiple measurements; for PCPT and SELECT, we include results using the first measurement.
We also used two sets of variants to calculate the PRSs in each of the two sets of individuals. We first utilized the independent genome-wide significant variants discovered in our analyses (one for discovery and one for the meta-analysis of discovery and replication). Second, we constructed a genome-wide score using PRS-CSx v2023-08-10 (ref. 75), which was implemented utilizing GWAS summary statistics, the 1,287,078 HapMap3 variants as an LD reference that had an imputation quality ≥0.9 in SELECT, and a global shrinkage parameter of ϕ = 0.0001 (which performed well in our previous work19). Because PRS-CSx only considers autosomes, independent genome-wide significant X chromosome variants were included (and produced a negligible increase in performance). The final scores were calculated by summing the effect size times the (probabilistic) number of alleles at each locus with PLINK v2.00a3.7LM76.
We also assessed the variance explained by the discovery PRS-CSx within age intervals in GERA; we looked only in GERA to have an out-of-sample estimate from discovery and a large enough sample size at each age. An individual could be in multiple bins, but we used just the first measurement of that individual per age bin.
Genetic adjustment of PSA for prostate cancer screening in GERA
We adjusted PSA as described previously19. Briefly, PSA values for individual i were adjusted by PSAiadj = PSAi/ai, where ai is a personalized adjustment factor derived from our PRS, as: ai = exp(PRSi)/exp(mean(PRS)). Here we estimated the mean(PRS) value within each group in GERA. We then evaluated the potential utility to alter biopsy referrals using age-specific PSA thresholds used within the Kaiser system (40–49 years = 2.5, 50–59 years = 3.5, 60–69 years = 4.5, and 70–79 years = 6.5 ng ml−1 (ref. 77)), evaluating net reclassification in cases and controls19.
We also tested for associations of our PSAadj with Gleason score (≤6, 7 and ≥8) using multinomial logistic regression with the R (ref. 45) v4.2.0 package nnet v7.3.18 (ref. 78).
To assess whether there was variability in PSA adjustment across PSA levels, we first binned PSA values (with narrower ranges for lower values where there was more data). Within each bin, we computed PSA − PSAadjusted, and then computed the median and IQR of these values. The median and IQR were then plotted at the center point of each bin by adding them to the identity line.
Genetically adjusted midlife PSA prostate cancer prediction impact
We next investigated the impact of genetically adjusting PSA on the prediction of overall and aggressive prostate cancer in GERA (3,540 cases (1,028 aggressive, Gleason ≥7), 21,702 controls). We constructed a midlife PSA25 based on each participant’s median PSA between 50 and 60 years, with cases restricted to measurements ≥1 year before diagnosis. Genetic PSA adjustment was performed as in the previous section. Associations between PSA or genetically adjusted ln PSA and prostate cancer risk were assessed using logistic regression for overall prostate cancer cases vs controls and for aggressive cases vs. controls, adjusting for covariates in Supplementary Table 1. Area under the curve was estimated using 10-fold repeated cross-validation (10 repeats) with caret v6.0.90 (ref. 79).
Bias-corrected prostate cancer estimates
Prostate cancer associations in individuals with EUR in the PRACTICAL consortium26 were adjusted for screening bias27, using estimates previously derived19: β’Cancer = βCancer − bβPSA, SE’Cancer = sqrt(SECancer2 + b2SEPSA2 + SEb2βPSA2 + SEb2SEPSA2), where SE is the standard error, and estimates were b = 1.144, and SEb = 2.909 × 10−4.
Associations with urinary symptom variants
We evaluated whether the novel PSA variants were associated with urinary symptoms in GERA, where participants completed the first 6 (of 7) questions from the American Urological Association Symptom Index (AUA-SI)80 with 5-point Likert scale responses. The questions asked about incomplete emptying, frequency, intermittency, urgency, weak stream and straining (Supplementary Table 13). The one missing question from the AUA-SI regarded nocturia. We calculated total scores as the sum of the questions, giving each individual a value ranging from 6 to 30. The score was dichotomized at <7, ≥7 to differentiate men with little or no BPH (n = 12,846) from those with moderate or severe BPH (n = 15,480). We then assessed the association between the PSA variants and the urinary symptom score.
Prostate volume analysis
We evaluated associations between PSA variants and prostate volume in patients on active surveillance (AS) enrolled in the Canary PASS. Between 2008 and 2017, Canary PASS prospectively enrolled 1,455 patients with clinically localized prostate cancer (cT1-cT2 and Gleason Grade 1–2) to undergo AS at 1 of 10 national sites81. Prostate volume was measured at diagnosis, with a median measurement of 43.0cc (IQR = 31.0–57.5). The median age at diagnosis was 63 years (IQR = 58–67), and 85% of Canary PASS self-reported as EUR. Genotyping was conducted in 1,220 participants82. We assessed potential associations between the 407 PSA variants that we successfully imputed in Canary PASS and prostate volume using mixed models with fixed effects for genetic variants, age at diagnosis, and 10 PCs, and a random effect for a genetic relationship matrix.
Associations with KLK3 plasma pQTL and eGenes
We annotated the 447 variants from the joint meta-analysis using recently published plasma pQTL association results for KLK3 from the UKB Pharma Proteomics Project using the Olink Explore platform29. We also used single-cell RNA sequencing data to assess whether the eGenes for PSA-associated variants (Supplementary Table 7) are expressed in secretory prostate cell types (particularly luminal epithelial cells) more than other prostate cell types (n = 36 cell types with >1,000 cells; 78,613 cells with eGenes total). For these analyses, we used data from the Chan-Zuckerberg Cell by Gene census v2023-12-15 (ref. 83).
Statistics and reproducibility
No statistical method was used to predetermine sample size, as all available samples were used to maximize power. Some analysis excluded individuals at a high risk of undiagnosed prostate cancer, as described above. Otherwise data were not excluded from the analysis. This study used only observational data (randomization and blinding are inapplicable).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Summary statistics and PRS weights (PRS318, PRS447, PRSCSx,disc, PRSCSx,joint) are available from the GWAS catalog (https://www.ebi.ac.uk/gwas/) under accession code GCST90461907, and PRS weights are available from the PRS catalog (https://www.pgscatalog.org/) under publication ID PGP000692 and score IDs PGS005098-PGS005101. Data from several studies are available on dbGaP under accession codes phs001391.v1.p1 (SCSS, PCPT, SELECT, Uganda, AAPC) and phs000306.v4.p1 (MEC). To protect individuals’ privacy, the following datasets are subject to controlled access: MVP data are available to Veterans Affairs researchers (https://www.mvp.va.gov/pwa/mvp-data-available-research), and, although opportunities for accessing MVP data are evolving, are currently limited to Veterans Affairs affiliated researchers; BioMe data are available by application and approval (https://icahn.mssm.edu/research/ipm/programs/biome-biobank/researcher-faqs); MADCaP data are available by application and approval (https://www.madcapnetwork.org/dataaccess); COMPASS data are available by application and approval (https://uwaterloo.ca/compass-system/information-researchers); GERA data are available upon approved applications to the Kaiser Permanente Research Bank Portal (https://researchbank.kaiserpermanente.org/for-researchers); UKB data are available in the UKB cloud-based Research Analysis Platform (https://www.ukbiobank.ac.uk). GTEx data were obtained from the GTEx portal (www.gtexportal.org) and can be obtained from dbGaP accession phs000424.v8.p2. Genome-wide summary statistics for the replication are available from https://doi.org/10.5281/zenodo.7460134 (ref. 84).
Code availability
Meta-analyses were conducted with a custom-patched METAL v2011-03-25 (https://csg.sph.umich.edu/abecasis/metal/download/)67 that prevents numerical precision loss (lines 633 and 635 of ‘Main.cpp’ modified to the number 15 to output 15 digits precision). All other analyses used unmodified publicly available software, as follows. Genome-wide association analyses were conducted using PLINK v2.00a3.7LM (http://www.cog-genomics.org/plink/2.0/)76. Additional meta-analyses were conducted with mJAM v2022-08-05 (https://github.com/USCbiostats/hJAM)20. Imputation was done via imputation servers (Michigan imputation server v1.5.7, https://imputationserver.sph.umich.edu (ref. 58), with Minimac4 v1.0.2, https://github.com/statgen/Minimac4 (ref. 59), and Eagle v2.4, https://alkesgroup.broadinstitute.org/Eagle/ (ref. 60)), Minimac3 v2.0.1 (https://genome.sph.umich.edu/wiki/Minimac3)59, and IMPUTE2 v2.3.2 (https://mathgen.stats.ox.ac.uk/impute/impute_v2.html)61. Analyses were also conducted in R, including v4.2.0 (https://cran.r-project.org/)45, with packages nnet v7.3.18 (ref. 78), and caret v6.0.90 (ref. 79). FUMA v1.5.2 (https://fuma.ctglab.nl)69, GTeX v8 (www.gtexportal.org)69, CADD v1.0 (https://cadd.bihealth.org/)60 and RefSeq v226 (https://www.ncbi.nlm.nih.gov/refseq/)71 were used for annotation. Circos plots were generated using Circos v0.69-6 (ref. 72). The genome-wide PRS was conducted with PRS-CSx v2023-08-10 (https://github.com/getian107/PRScsx)75.
References
Lilja, H. A kallikrein-like serine protease in prostatic fluid cleaves the predominant seminal vesicle protein. J. Clin. Invest. 76, 1899–1903 (1985).
Lilja, H., Ulmert, D. & Vickers, A. J. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nat. Rev. Cancer 8, 268–278 (2008).
Balk, S. P., Ko, Y.-J. & Bubley, G. J. Biology of prostate-specific antigen. J. Clin. Oncol. 21, 383–391 (2003).
Pinsky, P. F. et al. Prostate volume and prostate-specific antigen levels in men enrolled in a large screening trial. Urology 68, 352–356 (2006).
Lee, S. E. et al. Relationship of prostate-specific antigen and prostate volume in Korean men with biopsy-proven benign prostatic hyperplasia. Urology 71, 395–398 (2008).
Grubb, R. L. et al. Serum prostate-specific antigen hemodilution among obese men undergoing screening in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Cancer Epidemiol. Biomarkers Prev. 18, 748–751 (2009).
Harrison, S. et al. Systematic review and meta-analysis of the associations between body mass index, prostate cancer, advanced prostate cancer, and prostate-specific antigen. Cancer Causes Control 31, 431–449 (2020).
Jemal, A. et al. Prostate cancer incidence and PSA testing patterns in relation to USPSTF screening recommendations. JAMA 314, 2054–2061 (2015).
Gulati, R., Inoue, L. Y. T., Gore, J. L., Katcher, J. & Etzioni, R. Individualized estimates of overdiagnosis in screen-detected prostate cancer. J. Natl Cancer Inst. 106, djt367 (2014).
Sammon, J. D. et al. Prostate-specific antigen screening after 2012 US preventive services task force recommendations. JAMA 314, 2077–2079 (2015).
Hugosson, J. et al. A 16-yr follow-up of the european randomized study of screening for prostate cancer. Eur. Urol. 76, 43–51 (2019).
Sandhu, G. S. & Andriole, G. L. Overdiagnosis of prostate cancer. J. Natl Cancer Inst. Monogr. 2012, 146–151 (2012).
Frånlund, M. et al. Results from 22 years of followup in the Göteborg randomized population-based prostate cancer screening trial. J. Urol. 208, 292–300 (2022).
US Preventive Services Task Force. Screening for Prostate Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 319, 1901–1913 (2018).
Bell, N. et al. Recommendations on screening for prostate cancer with the prostate-specific antigen test. CMAJ 186, 1225–1234 (2014).
Tikkinen, K. A. O. et al. Prostate cancer screening with prostate-specific antigen (PSA) test: a clinical practice guideline. Br. Med. J. 362, k3581 (2018).
Bansal, A. et al. Heritability of prostate-specific antigen and relationship with zonal prostate volumes in aging twins. J. Clin. Endocrinol. Metab. 85, 1272–1276 (2000).
Pilia, G. et al. Heritability of cardiovascular and personality traits in 6,148 Sardinians. PLoS Genet. 2, e132 (2006).
Kachuri, L. et al. Genetically adjusted PSA levels for prostate cancer screening. Nat. Med. 29, 1412–1423 (2023).
Shen, J. et al. Hierarchical joint analysis of marginal summary statistics—Part I: Multipopulation fine mapping and credible set construction. Genet. Epidemiol. 48, 241–257 (2024).
Kerem, B. S. et al. DNA marker haplotype association with pancreatic sufficiency in cystic fibrosis. Am. J. Hum. Genet. 44, 827–834 (1989).
Lippman, S. M. et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 301, 39–51 (2009).
Thompson, I. M. et al. Assessing prostate cancer risk: results from the Prostate Cancer Prevention Trial. J. Natl Cancer Inst. 98, 529–534 (2006).
Mayo, K. R. et al. The all of us data and research center: creating a secure, scalable, and sustainable ecosystem for biomedical research. Annu Rev. Biomed. Data Sci. 6, 443–464 (2023).
Preston, M. A. et al. Baseline prostate-specific antigen levels in midlife predict lethal prostate cancer. J. Clin. Oncol. 34, 2705–2711 (2016).
Conti, D. V. et al. Trans-ancestry genome-wide association meta-analysis of prostate cancer identifies new susceptibility loci and informs genetic risk prediction. Nat. Genet. 53, 65–75 (2021).
Dudbridge, F. et al. Adjustment for index event bias in genome-wide association studies of subsequent events. Nat. Commun. 10, 1561 (2019).
Jiang, L., Zheng, Z., Fang, H. & Yang, J. A generalized linear mixed model association tool for biobank-scale data. Nat. Genet. 53, 1616–1621 (2021).
Eldjarn, G. H. et al. Large-scale plasma proteomics comparisons through genetics and disease associations. Nature 622, 348–358 (2023).
Kim, J. & Coetzee, G. A. Prostate specific antigen gene regulation by androgen receptor. J. Cell. Biochem. 93, 233–241 (2004).
Heinlein, C. A. & Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 25, 276–308 (2004).
Agúndez, J. A. G. et al. The diamine oxidase gene is associated with hypersensitivity response to non-steroidal anti-inflammatory drugs. PLoS ONE 7, e47571 (2012).
Amo, G. et al. FCERI and histamine metabolism gene variability in selective responders to NSAIDS. Front Pharm. 7, 353 (2016).
Miller, C. L. et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun. 7, 12092 (2016).
Linseman, T. et al. Functional validation of a common nonsynonymous coding variant in ZC3HC1 associated with protection from coronary artery disease. Circ. Cardiovasc. Genet. 10, e001498 (2017).
Cuppens, H. & Cassiman, J.-J. CFTR mutations and polymorphisms in male infertility. Int. J. Androl. 27, 251–256 (2004).
Landrum, M. J. et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 48, D835–D844 (2020).
Kawa, S. et al. Azoospermia in mice with targeted disruption of the Brek/Lmtk2 (brain-enriched kinase/lemur tyrosine kinase 2) gene. Proc. Natl Acad. Sci. USA 103, 19344–19349 (2006).
Cruz, D. F., Farinha, C. M. & Swiatecka-Urban, A. Unraveling the function of lemur tyrosine kinase 2 network. Front. Pharmacol. 10, 24 (2019).
Bostwick, D. G. et al. The association of benign prostatic hyperplasia and cancer of the prostate. Cancer 70, 291–301 (1992).
Ørsted, D. D. & Bojesen, S. E. The link between benign prostatic hyperplasia and prostate cancer. Nat. Rev. Urol. 10, 49–54 (2013).
Thompson, I. M. et al. Prevalence of prostate cancer among men with a prostate-specific antigen level < or =4.0 ng per milliliter. N. Engl. J. Med. 350, 2239–2246 (2004).
Coric, J., Mujic, J., Kucukalic, E. & Ler, D. Prostate-specific antigen (PSA) and prostate volume: better predictor of prostate cancer for Bosnian and Herzegovina men. Open Biochem. J. 9, 34–36 (2015).
D’Amico, A. V. Risk-based management of prostate cancer. N. Engl. J. Med. 365, 169–171 (2011).
R Core Team R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2012).
McGrath, S., Zhao, X., Qin, Z. Z., Steele, R. & Benedetti, A. One-sample aggregate data meta-analysis of medians. Stat. Med. 38, 969–984 (2019).
Tayo, B. O. et al. Genetic background of patients from a university medical center in Manhattan: implications for personalized medicine. PLoS ONE 6, e19166 (2011).
Aschebrook-Kilfoy, B. et al. Cohort profile: the ChicagO Multiethnic Prevention and Surveillance Study (COMPASS). BMJ Open 10, e038481 (2020).
Press, D. J. et al. Tobacco and marijuana use and their association with serum prostate-specific antigen levels among African American men in Chicago. Prev. Med. Rep. 20, 101174 (2020).
Andrews, C. et al. Development, evaluation, and implementation of a pan-African cancer research network: men of African descent and carcinoma of the prostate. J. Glob. Oncol. 4, 1–14 (2018).
Harlemon, M. et al. A custom genotyping array reveals population-level heterogeneity for the genetic risks of prostate cancer and other cancers in Africa. Cancer Res. 80, 2956–2966 (2020).
Kolonel, L. N., Altshuler, D. & Henderson, B. E. The multiethnic cohort study: exploring genes, lifestyle and cancer risk. Nat. Rev. Cancer 4, 519–527 (2004).
Kolonel, L. N. et al. A multiethnic cohort in Hawaii and Los Angeles: baseline characteristics. Am. J. Epidemiol. 151, 346–357 (2000).
Gaziano, J. M. et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 70, 214–223 (2016).
Fang, H. et al. Harmonizing genetic ancestry and self-identified race/ethnicity in genome-wide association studies. Am. J. Hum. Genet. 105, 763–772 (2019).
Signorello, L. B. et al. Southern community cohort study: establishing a cohort to investigate health disparities. J. Natl Med. Assoc. 97, 972–979 (2005).
Venner, E. et al. The frequency of pathogenic variation in the All of Us cohort reveals ancestry-driven disparities. Commun. Biol. 7, 174 (2024).
Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
Loh, P.-R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Howie, B., Fuchsberger, C., Stephens, M., Marchini, J. & Abecasis, G. R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 44, 955–959 (2012).
1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Sheng, X. et al. Inverted genomic regions between reference genome builds in humans impact imputation accuracy and decrease the power of association testing. HGG Adv. 4, 100159 (2023).
Karolchik, D. et al. The UCSC Genome Browser database: 2014 update. Nucl. Acids Res. 42, D764–D770 (2014).
Ganesh, S. K. et al. Effects of long-term averaging of quantitative blood pressure traits on the detection of genetic associations. Am. J. Hum. Genet. 95, 49–65 (2014).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Hoffmann, T. J. et al. Genome-wide association study of prostate-specific antigen levels identifies novel loci independent of prostate cancer. Nat. Commun. 8, 14248 (2017).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745 (2016).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Gerstenbluth, R. E., Maniam, P. N., Corty, E. W. & Seftel, A. D. Prostate-specific antigen changes in hypogonadal men treated with testosterone replacement. J. Androl. 23, 922–926 (2002).
Guess, H. A., Heyse, J. F. & Gormley, G. J. The effect of finasteride on prostate-specific antigen in men with benign prostatic hyperplasia. Prostate 22, 31–37 (1993).
Ruan, Y. et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 54, 573–580 (2022).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Oesterling, J. E. et al. Serum prostate-specific antigen in a community-based population of healthy men. Establishment of age-specific reference ranges. JAMA 270, 860–864 (1993).
Ripley, W. N. & Ripley, B. D. Modern Applied Statistics with S (Springer, 2002).
Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 28, 1–26 (2008).
Barry, M. J. et al. The American Urological Association Symptom Index for benign prostatic hyperplasia. J. Urol. 197, S189–S197 (2017).
Newcomb, L. F. et al. Canary Prostate Active Surveillance Study: design of a multi-institutional active surveillance cohort and biorepository. Urology 75, 407–413 (2010).
Jiang, Y. et al. Genetic factors associated with prostate cancer conversion from active surveillance to treatment. HGG Adv. 3, 100070 (2022).
Program, C. S.-C. B. et al. CZ CELL×GENE Discover: a single-cell data platform for scalable exploration, analysis and modeling of aggregated data. Nucleic Acids Res. 53, D886–D900 (2025).
Linda, K. et al. Genetically adjusted PSA levels for prostate cancer screening. Zenodo https://doi.org/10.5281/zenodo.7460134 (2022).
Acknowledgements
We thank the participants who participated in each cohort. This research is based in part on data from the MVP, Office of Research and Development, Veterans Health Administration, and was supported by the MVP017 Exemplar Cancer Project. This research was conducted using the UKB resource under application #14105.
The Precision PSA study is supported by funding from the NIH National Cancer Institute (NCI) under award numbers R01CA241410 (PI: J.S.W.) and U01CA261339 (MPI: J.S.W.). L.K. is supported by funding from NIH/NCI (R00CA246076). R.E.G. is supported by a Young Investigator Award from the Prostate Cancer Foundation. H.L. is supported in part by NIH/NCI by a Cancer Center Support Grant to Memorial Sloan Kettering Cancer Center (P30 CA008748, PI: S. Vickers), U01-CA266535 (PI: S. Carlsson), R01-CA244948 (PI: R.J.K.), and Swedish Cancer Society (Cancerfonden 20 1354 PJF; PI: H.L.). This work was supported by research grants from the NIH National Institute of General Medical Sciences under award number R01GM130791 (PI: J.D.M.); the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai; the Office of Research Infrastructure of the NIH under award number S10OD026880 and NIH/NCI funding (R01CA175491, R01CA244948; PI: R.J.K.); the NCI/NIH (UM1CA182883, PI: C. M. Tangen/I. M. Thompson; U10CA37429, PI: C. D. Blanke). MADCaP was supported by U01CA184374 (PI: T.R.). COMPASS was supported by P30CA014599. Support for GERA participant enrollment, survey completion and biospecimen collection for RPGEH was provided by the Robert Wood Johnson Foundation, the Wayne and Gladys Valley Foundation, the Ellison Medical Foundation and Kaiser Permanente national and regional benefit programs. GERA genotyping was funded by National Institute on Aging and NIH Common Fund (grant RC2 AG-036607 to C. Schaeffer and N. Risch). The All of Us Research Program is supported by the NIH, Office of the Director: Regional Medical Centers: 1 OT2 OD026549; 1 OT2 OD026554; 1 OT2 OD026557; 1 OT2 OD026556; 1 OT2 OD026550; 1 OT2 OD 026552; 1 OT2 OD026553; 1 OT2 OD026548; 1 OT2 OD026551; 1 OT2 OD026555; IAA #AOD 16037; Federally Qualified Health Centers: HHSN 263201600085U; Data and Research Center: 5 U2C OD023196; Biobank: 1 U24 OD023121; The Participant Center: U24 OD023176; Participant Technology Systems Center: 1 U24 OD023163; Communications and Engagement: 3 OT2 OD023205; 3 OT2 OD023206; and Community Partners: 1 OT2 OD025277; 3 OT2 OD025315; 1 OT2 OD025337; 1 OT2 OD025276.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and analysis, the decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
T.J.H., R.E.G., L.K. and J.S.W. contributed to the study concept and design. T.J.H., R.E.G., R.K.M., A.A.R., C.L.C., K.F., Y.J., A.W., J.D.M., D.V.C., L.K. and J.S.W. were responsible for the acquisition, analysis or interpretation of data. T.J.H., R.E.G., L.K. and J.S.W. drafted the paper. T.J.H., R.E.G., R.K.M., A.A.R., C.L.C., K.F., Y.J., A.W., R.J.K., B.L.P., S.E., L.T., W.B., J.L., L.B.G., B.F.D., T.R., J.L., C.A., A.O.A., B.A., O.I.A.-S., P.W.F., M.J., R.J., W.C.C., J.E.M., I.A., S.I.B., J.P.S., K.S., M.J.M., N.D.F., W.-Y.H., S.A.L., P.J.G., C.T., I.T., H.L., D.K.R., J.P., S.K.V.D.E., S.J.C., J.D.M., D.V.C., C.A.H., A.C.J., L.K. and J.S.W. critically revised the paper for important intellectual content.
Corresponding authors
Ethics declarations
Competing interests
J.S.W. and C.L.C. are nonemployee co-founders of Avail Bio. H.L. is named on a patent for intact PSA assays and a patent for a statistical method to detect prostate cancer that is licensed to and commercialized by OPKO Health. H.L. receives royalties from sales of the test and has stock in OPKO Health. J.S.W. consults for DLA Piper on subject matter unrelated to this study. R.E.G. consults for Hunton Andrews Kurth on subject matter unrelated to this study. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Brian Helfand, Tyler Seibert and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Overlap of SNPs in groups.
a, Discovery (n = 296,754), genome-wide significant. b, Discovery, Bonferroni significant. c, Meta-analysis (n = 95,768), genome-wide significant. d, Meta-analysis, Bonferroni significant. e, Previously reported variants, genome-wide significant. f, Previously reported variants, Bonferroni significant. SNPs were identified from fixed-effects meta-analysis of linear regression tests, two-sided. EUR, European ancestry; AFR, African ancestry; ASN, Asian ancestry; LAT, Hispanic/Latino.
Extended Data Fig. 2 Medication sensitivity analysis.
Comparison of effect estimates from the main UK Biobank European ancestry PSA analyses to those from the sensitivity analyses excluding individuals taking testosterone or 5-alpha reductase inhibitors, which are medications that could affect PSA levels. We also adjusted for alpha-blockers in this analysis. Results are from linear regression tests (n = 26,669, nsensitivity = 20,742), two-sided, no multiple comparison adjustment. PSA, prostate-specific antigen.
Extended Data Fig. 3 Biopsy reclassification with genetically adjusted PSA in additional groups.
a, b, PSA levels were adjusted (see Methods) using the PRS-CSx estimate from the out-of-sample discovery cohort, assessed in GERA using age-specific cutoffs in (a) Hispanic/Latino (n = 403) and (b) East Asian ancestry (n = 406). The sankey diagram is based on percentages of each of the flows from/into nodes.
Extended Data Fig. 4 Measured PSA vs. genetically adjusted PSA (PSA’).
Comparison given using the Kaiser Permanente GERA cohort (n = 43,945). The variability is consistent across ln PSA levels; solid black line identifies the median, and the dashed line the interquartile range (adjustment described in Methods). GERA, Genetic Epidemiology Resource on Adult health and aging.
Extended Data Fig. 5 Comparison of genetic effects on absolute and relative quantification of PSA.
Comparison done in UKB using Olink. a, b, Correlation between multiancestry GWAS effect sizes (from fixed effects meta-analysis of linear regressions) for 409 out of 447 lead variants and KLK3 pQTL effect sizes was estimated in (a) European (n = 46,214) and (b) African (n = 1,065) ancestry populations. Error bars are ± standard errors. UKB, UK Biobank.
Extended Data Fig. 6 Prostate cell expression.
Expression of eQTL genes (eGenes) associated with discovered SNPs is shown for prostate cell types (scRNA-seq Chan-Zuckerberg BioHub, n = 36 cell types with >1,000 cells; 78,613 cells with eGenes total). Circle size indicates the fraction of cells with expression, and expression levels are colored. Expression across all eQTL genes except LIME1 is observed in luminal cells of the prostate.
Extended Data Fig. 7 Luminal epithelium vs. all other prostate cells expression.
Expression of eQTL-associated genes (eGenes) is represented as a percentile of total expression in luminal epithelium (n = 14,380 cells) vs. all other prostate tissue cells (n = 68,240 cells). eQTL-associated genes are expressed at significantly higher percentile levels in luminal epithelial cells than other prostate cell types (P = 0.0006).
Supplementary information
Supplementary Information
Supplementary Fig. 1.
Supplementary Tables
Supplementary Tables 1–17.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hoffmann, T.J., Graff, R.E., Madduri, R.K. et al. Genome-wide association study of prostate-specific antigen levels in 392,522 men identifies new loci and improves prediction across ancestry groups. Nat Genet 57, 334–344 (2025). https://doi.org/10.1038/s41588-024-02068-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-024-02068-z
