
Abstract
Grapevine (Vitis) is one of the oldest domesticated fruit crops with great cultural and economic importance. Here we assembled and annotated haplotype-resolved genomes of 72 global Vitis accessions including 25 wild and 47 cultivated grapevines, among which genomes for 60 grapevines are newly released. Haplotype-aware phylogenomics disentangled the mysterious hybridization history of grapevines, revealing the enormous genetic diversity of the Vitis genus. Pangenomic analysis reveals that European cultivars, more susceptible to the destructive disease downy mildew (DM), have a smaller repertoire of resistance genes in the NLR family encoding the TIR-NBARC-LRR ___domain. Through extensive structural variation (SV) characterization, phenotyping, DM-infection transcriptome profiling of 113 Vitis accessions, and SV–expression quantitative trait loci analysis, we have identified over 63 SVs and their relevant genes significantly associated with DM resistance, exemplified by a lysine histidine transporter, VvLHT8. This haplotype-resolved super pangenome of the Vitis genus will accelerate breeding and enrich our understanding of the evolution and biology of grapevines.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
209,00 € per year
only 17,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
The genome sequencing, ChIP–seq and RNA-seq data that support the findings of this study have been deposited in the NCBI with the BioProject accession PRJNA1021789. The assemblies and annotations have been deposited in Zendo105 (https://doi.org/10.5281/zenodo.13982444). The assemblies have also been deposited in the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/), European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/home) and National Genomics Data Center (https://ngdc.cncb.ac.cn; see Supplementary Table 16 for accession numbers). Source data are provided with this paper.
Code availability
All code and software sources used in our paper are listed in the Methods, along with corresponding references. Other customized codes we used have been deposited in GitHub (https://github.com/NotebookOFXiaoMing/SuperPangenomeofGrapevines) and Zenodo105 (https://doi.org/10.5281/zenodo.13982444).
References
Dong, Y. et al. Dual domestications and origin of traits in grapevine evolution. Science 379, 892–901 (2023).
Aradhya, M. K. et al. Genetic structure and differentiation in cultivated grape, Vitis vinifera L. Genet. Res. 81, 179–192 (2003).
Terral, J.-F. et al. Evolution and history of grapevine (Vitis vinifera) under domestication: new morphometric perspectives to understand seed domestication syndrome and reveal origins of ancient European cultivars. Ann. Bot. 105, 443–455 (2010).
Grassi, F. & De Lorenzis, G. Back to the origins: background and perspectives of grapevine domestication. Int. J. Mol. Sci. 22, 4518 (2021).
Cantu, D. & Walker, M. A. (eds). The Grape Genome pp. 25–38 (Springer International Publishing, 2019).
Liang, Z. et al. Whole-genome resequencing of 472 Vitis accessions for grapevine diversity and demographic history analyses. Nat. Commun. 10, 1190 (2019).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Zhou, Y. et al. The population genetics of structural variants in grapevine domestication. Nat. Plants 5, 965–979 (2019).
Shirasawa, K. et al. De novo whole-genome assembly in an interspecific hybrid table grape, ‘Shine Muscat. DNA Res. 29, dsac040 (2022).
Velt, A. et al. An improved reference of the grapevine genome reasserts the origin of the PN40024 highly homozygous genotype. G3 (Bethesda) 13, jkad067 (2023).
Shi, X. et al. The complete reference genome for grapevine (Vitis vinifera L.) genetics and breeding. Hortic. Res. 10, uhad061 (2023).
Zhang, K. et al. The haplotype-resolved T2T genome of teinturier cultivar Yan73 reveals the genetic basis of anthocyanin biosynthesis in grapes. Hortic. Res. 10, uhad205 (2023).
Wang, X. Telomere-to-telomere and gap-free genome assembly of a susceptible grapevine species (Thompson Seedless) to facilitate grape functional genomics. Hortic. Res. 11, uhad260 (2023).
Danilevicz, M. F., Tay Fernandez, C. G., Marsh, J. I., Bayer, P. E. & Edwards, D. Plant pangenomics: approaches, applications and advancements. Curr. Opin. Plant Biol. 54, 18–25 (2020).
Liao, W.-W. et al. A draft human pangenome reference. Nature 617, 312–324 (2023).
Zhou, Y. et al. Assembly of a pangenome for global cattle reveals missing sequences and novel structural variations, providing new insights into their diversity and evolutionary history. Genome Res. 32, 1585–1601 (2022).
Jiang, Y.-F. et al. Pangenome obtained by long-read sequencing of 11 genomes reveal hidden functional structural variants in pigs. iScience 26, 106119 (2023).
Li, R. et al. A sheep pangenome reveals the spectrum of structural variations and their effects on tail phenotypes. Genome Res. 33, 463–477 (2023).
Liu, Y. et al. Pan-genome of wild and cultivated soybeans. Cell 182, 162–176.e13 (2020).
Shang, L. et al. A super pan-genomic landscape of rice. Cell Res. 32, 878–896 (2022).
Li, N. et al. Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species. Nat. Genet. 55, 852–860 (2023).
Zhou, Y. et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 606, 527–534 (2022).
Cochetel, N. et al. A super-pangenome of the North American wild grape species. Genome Biol. 24, 290 (2023).
Long, Q. et al. Population comparative genomics discovers gene gain and loss during grapevine domestication. Plant Physiol. 195, 1401–1413 (2024).
Liu, Z. et al. Grapevine pangenome facilitates trait genetics and genomic breeding. Nat. Genet. 56, 2804–2814 (2024).
Cheng, H. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 40, 1332–1335 (2022).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Cochetel, N. et al. Diploid chromosome-scale assembly of the Muscadinia rotundifolia genome supports chromosome fusion and disease resistance gene expansion during Vitis and Muscadinia divergence. G3 (Bethesda) 11, jkab033 (2021).
Garrison, E. et al. Building pangenome graphs. Nat. Methods 21, 2008–2012 (2024).
Zhang, C., Scornavacca, C., Molloy, E. K. & Mirarab, S. ASTRAL-Pro: quartet-based species-tree inference despite paralogy. Mol. Biol. Evol. 37, 3292–3307 (2020).
Marone, D., Russo, M., Laidò, G., De Leonardis, A. & Mastrangelo, A. Plant nucleotide binding site-leucine-rich repeat (NBS-LRR) genes: active guardians in host defense responses. Int. J. Mol. Sci. 14, 7302–7326 (2013).
Steuernagel, B. et al. The NLR-annotator tool enables annotation of the intracellular immune receptor repertoire. Plant Physiol. 183, 468–482 (2020).
Feechan, A. et al. Genetic dissection of a TIR-NB-LRR locus from the wild North American grapevine species Muscadinia rotundifolia identifies paralogous genes conferring resistance to major fungal and oomycete pathogens in cultivated grapevine. Plant J. 76, 661–674 (2013).
Foria, S. et al. Gene duplication and transposition of mobile elements drive evolution of the Rpv3 resistance locus in grapevine. Plant J. 101, 529–542 (2020).
Jones, J. D. G., Staskawicz, B. J. & Dangl, J. L. The plant immune system: From discovery to deployment. Cell 187, 2095–2116 (2024).
Goel, M., Sun, H., Jiao, W.-B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 277 (2019).
Foria, S. et al. InDel markers for monitoring the introgression of downy mildew resistance from wild relatives into grape varieties. Mol. Breed. 38, 124 (2018).
Di Gaspero, G. et al. Selective sweep at the Rpv3 locus during grapevine breeding for downy mildew resistance. Theor. Appl. Genet. 124, 277–286 (2012).
Zhou, H. et al. Patellin protein family functions in plant development and stress response. J. Plant Physiol. 234–235, 94–97 (2019).
He, Q. et al. A graph-based genome and pan-genome variation of the model plant Setaria. Nat. Genet. 55, 1232–1242 (2023).
Sirén, J. et al. Pangenomics enables genotyping of known structural variants in 5202 diverse genomes. Science 374, abg8871 (2021).
GTEx Consortium. et al. The impact of structural variation on human gene expression. Nat. Genet. 49, 692–699 (2017).
Liu, G. et al. Amino acid homeostasis modulates salicylic acid-associated redox status and defense responses in Arabidopsis. Plant Cell 22, 3845–3863 (2010).
Murata, Y., Mori, I. C. & Munemasa, S. Diverse stomatal signaling and the signal integration mechanism. Annu. Rev. Plant Biol. 66, 369–392 (2015).
Coupel-Ledru, A. et al. Reduced nighttime transpiration is a relevant breeding target for high water-use efficiency in grapevine. Proc. Natl Acad. Sci. USA 113, 8963–8968 (2016).
Ye, W. et al. Stomatal immunity against fungal invasion comprises not only chitin-induced stomatal closure but also chitosan-induced guard cell death. Proc. Natl Acad. Sci. USA 117, 20932–20942 (2020).
Cantu, D. & Walker, M. A. (eds). The Grape Genome pp. 1–24 (Springer International Publishing, 2019).
Minio, A., Cochetel, N., Massonnet, M., Figueroa-Balderas, R. & Cantu, D. HiFi chromosome-scale diploid assemblies of the grape rootstocks 110R, Kober 5BB, and 101–14 Mgt. Sci. Data 9, 660 (2022).
Zou, C. et al. Multiple independent recombinations led to hermaphroditism in grapevine. Proc. Natl Acad. Sci. USA 118, e2023548118 (2021).
Massonnet, M. et al. The genetic basis of sex determination in grapes. Nat. Commun. 11, 2902 (2020).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Report. 15, 8–15 (1997).
Belton, J.-M. et al. Hi–C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Xu, M. et al. TGS-GapCloser: a fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience 9, giaa094 (2020).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 19, 705–710 (2022).
Poplin, R. et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983–987 (2018).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: assessing genomic data quality and beyond. Curr. Protoc. 1, e323 (2021).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457 (2020).
Nishimura, D. RepeatMasker. Biotech Softw. Internet Rep. 1, 36–39 (2000).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3, lqaa108 (2021).
Johnson, A. D. et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24, 2938–2939 (2008).
Cantarel, B. L. et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Wlodzimierz, P., Hong, M. & Henderson, I. R. TRASH: Tandem repeat annotation and structural hierarchy. Bioinformatics 39, btad308 (2023).
Huang, H.-R. et al. Telomere-to-telomere haplotype-resolved reference genome reveals subgenome divergence and disease resistance in triploid Cavendish banana. Hortic. Res. 10, uhad153 (2023).
Vollger, M. R., Kerpedjiev, P., Phillippy, A. M. & Eichler, E. E. StainedGlass: interactive visualization of massive tandem repeat structures with identity heatmaps. Bioinformatics 38, 2049–2051 (2022).
Chen, W. et al. Two telomere-to-telomere gapless genomes reveal insights into Capsicum evolution and capsaicinoid biosynthesis. Nat. Commun. 15, 4295 (2024).
Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Zhang, Y. et al. Model-based Analysis of ChIP–seq (MACS). Genome Biol. 9, R137 (2008).
Li, Q., Brown, J. B., Huang, H. & Bickel, P. J. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 5, 1752–1779 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.1–11.10.33 (2013).
Jeffares, D. C. et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 8, 14061 (2017).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Wu, T. et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Zhang, Z. et al. ParaAT: a parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 419, 779–781 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Hickey, G. et al. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 21, 35 (2020).
Shabalin, A. A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358 (2012).
Li, M.-X., Yeung, J. M. Y., Cherny, S. S. & Sham, P. C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756 (2012).
Zhang, M. et al. Plasma membrane H+-ATPase overexpression increases rice yield via simultaneous enhancement of nutrient uptake and photosynthesis. Nat. Commun. 12, 735 (2021).
Yao, X., Xia, N., Meng, X., Duan, C. & Pan, Q. A one-step polyphenol removal approach for detection of multiple phytohormones from grape Berry. Horticulturae 8, 548 (2022).
Yan, M., Ayhan, D. H. & Wang, X. Super pangenome of Vitis empowers identification of downy mildew resistance genes for grapevine improvement. Zenodo https://doi.org/10.5281/zenodo.13982444 (2024).
Acknowledgements
We thank the Bioinformatics Platform at Peking University Institute of Advanced Agricultural Sciences (PKU-IAAS) for providing high-performance computing resources. We thank X. Liu and Y. Li from PKU-IAAS Mass Spectrometry Platform for their technical support in the quantification of salicylic acid. Both W.Y. and L.G. are supported by the Key R&D Program of Shandong Province, China (2024CXPT031) and Shandong Provincial Natural Science Foundation (SYS202206), Taishan Scholars Program of Shandong Province and Weifang Key Laboratory of Grapevine Improvement and Utilization, China. L.G. is also supported by the Natural Science Foundation for Distinguished Young Scholars of Shandong Province, China (ZR2023JQ010).
Author information
Authors and Affiliations
Contributions
L.G. and W.Y. conceived and supervised the project. J.J., W.Z., J.M., L.L.G., X.Z., H.S. and C.L. curated and prepared the grapevine samples. D.W., W.Z., J.M., W.J., J.J., G.Q., L.L.G., Q.Y., X.Z., J.W. and H.S. performed the phenotyping and molecular experiments. D.M. and Q.Y. conducted ChIP–seq experiments. D.H.A., X.W., S.C., J.S. and S.Y. conducted genome assembly and annotation. D.H.A. and M.Y. performed variant calling, constructed pangenome graph and conducted eQTL mapping. L.G., D.H.A., M.S.R., X.W.D. and W.Y. interpreted results. M.N.B. provided technical assistance and participated in the discussion. L.G., D.H.A., M.S.R., X.W., M.Y. and W.Y. prepared the figures and tables and wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Sachiko Isobe and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Genome assembly and validation of haplotype-resolved complete genome of Vitis vinifera cv. Chardonnay.
a, Genome survey of winegrape cultivar Chardonnay based on k-mer frequency analysis. b, Chromatin interaction heatmap of two T2T haploid genomes using Hi-C data. c,d, The plot of LTR assembly index (LAI) for all chromosomes in two haploid genomes. e, Whole-genome sequence alignment of two T2T haploid genomes (horizontal) against a published genome PN40024 (vertical).
Extended Data Fig. 2 Centromere sequence alignment between haplotypes and among chromosomes.
a, Sequence identity heatmap of centromere sequence alignment across 19 chromosomes, showing the conservation and divergence of centromeres among chromosomes. b–d, Sequence identity heatmap of centromere sequence alignment between two haplotypes. Representative examples of three chromosomes are provided.
Extended Data Fig. 3 Population genomic analysis of grapevine accessions.
a, Box plot showing differences in stomatal density, length, and conductance in 71 accessions. For each boxplot, the box edges represent the interquartile range (IQR), with the centerline indicating the median. The whiskers extend to the most extreme data points within 1.5× IQR. b, Cross-validation error plot for k = 2–8 for 72 samples ADMIXTURE ancestry plot in Fig. 2f. c, ADMIXTURE ancestry plot for 591 samples based on SNPs. Each colored segment represents the proportion contributed by that ancestral population. d, Principal component analysis plot of 591 samples based on SNPs. A total of 71 accessions are shown in purple (Muscadine), blue (North American), green (East Asian) and red (European).
Extended Data Fig. 4 Haplotype-resolved genome assembly of 71 grapevine accessions.
a, Scatter plots showing two haplotypes of all 71 grapevine accessions for BUSCO, LAI, and QV values. Purple dots, Muscadine accessions; blue dots, North American accessions; red dots, European accessions; green dots, East Asian accessions. b, Box plot of chromosome size distributions of 71 grapevine accessions. For box plots, the central mark indicates the median, and the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considered outliers, and the outliers are plotted with ‘+’. c,d, Total length of transposable element contents (Mb) of different classes in 71 grapevine accessions with two-sided test. d, Two-sided t test. LTR: long terminal repeats, LINE: long interspersed nuclear elements, SINE: short interspersed nuclear elements, DNA: DNA transposons.
Extended Data Fig. 5 Pangenome analysis and functional enrichment.
a, Comparison of different groups of grapevine accessions for the content of softcore, dispensable and private genomes. b,c, Nucleotide diversity (b), dN/dS (c) of core, softcore, dispensable genes. a–c, Significance was tested using a Kruskal–Wallis test; multiple comparisons were analyzed using a Nemenyi test. The P values were not corrected for multiple comparisons. The different lowercase letters above the box plots represent significant differences (P ≤ 0.05). For each boxplot, the box edges represent the interquartile range (IQR), with the centerline indicating the median. The whiskers extend to the most extreme data points within 1.5× IQR. d–g, GO enrichment of pangenome core genes (d), softcore genes (e), private genes (f) and dispensable genes (g) for 144 haplotype grapevine genomes. The enriched GO categories were determined using the one-sided version of Fisher’s exact test, followed by the Benjamin–Hochberg correction to obtain adjusted P values for multiple testing.
Extended Data Fig. 6 Pan-nucleotide-binding site leucine-rich repeat (NLR) analysis.
a, Variation of NLR gene families in the pan- and core-NLR with additional grapevine genomes. At each given number of genomes, the number of combinations is 100. The lower and upper bounds of the box indicate the first and third quartiles, respectively, and the center line indicates the median. b, Compositions of NLR families in the grapevine pangenome and individual genome. The histogram shows the number of NLR gene families in the 144 haploid genomes with different frequencies where colors of histograms correspond to core, softcore, dispensable, and private NLR genes. The pie chart shows the proportion of the NLR gene family marked by each composition. c–l, The number of different NLR genes in Muscadine (n = 8), European (n = 43), North American (n = 43) and East Asian (n = 50) groups. Two-sided Student’s t test. For each boxplot, the box edges represent the interquartile range (IQR), with the centerline indicating the median. The whiskers extend to the most extreme data points within 1.5× IQR and the outliers are plotted with ‘black point’.
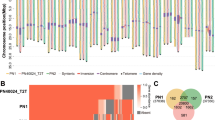
Extended Data Fig. 7 Structural variants in grapevines and Pan-SV analysis.
a, Distribution of inversion events in terms of their distance to centromeres. b, Distribution of translocation events in terms of their distance to centromeres. c–f, Examples of structural variants in grapevine accessions were identified both in whole-genome alignment (left) and Hi-C interaction heatmap (right). For each variant, the top is a reference vs. reference alignment and the bottom is a reference vs. sample alignment. Structural variants were marked using rectangles or dotted circles.
Extended Data Fig. 8 Expression and phenotype of UDV305 marker and nearby SVs.
a–c, Stomatal density, stomatal length, stomatal conductance at 10:00 AM and 10:00 PM of accessions with or without SVs on (a) and near (b,c) UDV305 marker. Purple: muscadine, blue: North American, green: East Asian, red: European accessions. *P < 0.05, n = 113, two-sided Wilcoxon rank sum test. For box plots, the central mark indicates the median, and the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considered outliers, and the outliers are of the 25th and 75th percentiles.
Extended Data Fig. 9 Expression and phenotype of UDV737 and INDEL-26032 markers and nearby SVs.
a,b, Gene expression during mock and P. viticola infection, sporangium number at 7 dpi, stomatal density, stomatal length, stomatal conductance at 10:00 AM and 10:00 PM of accessions with or without SVs near the marker UDV737 (a) and on the marker INDEL-26032 (b). Purple: muscadine, blue: North American, green: East Asian, red: European accessions. *P < 0.05, n = 113, two-sided Wilcoxon rank sum test. For box plots, the central mark indicates the median, and the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considered outliers, and the outliers are of the 25th and 75th percentiles.
Supplementary information
Supplementary Information
Supplementary Figs. 1–4.
Supplementary Tables 1–16
Supplementary Table 1. Summary statistics of telomeres assembled in V. vinifera cv. Chardonnay VHP-T2T genome. Supplementary Table 2. Characterization of centromere on each chromosome identified by CENH3 ChIP–seq. Supplementary Table 3. Information of 71 grapevine accessions used for de novo genome assembly. Supplementary Table 4. Stomatal traits and downy mildew infection phenotype of Vitis accessions used for transcriptome analysis. Supplementary Table 5. Summary statistics of genome sequencing reads (PacBio HiFi, NGS, ONT and Hi-C) generated for the 72 Vitis accessions. Supplementary Table 6. Predicted genome sizes for 71 accessions based on k-mer frequency analysis of short reads. Supplementary Table 7. Summary statistics for chromosome-level haplotype-resolved genome assembly, annotation and quality assessment for 71 grapevine accessions (142 haplotypes). Supplementary Table 8. The comparison of two phylogenetic trees for 71 grapevine accessions, constructed either using reference-based SNPs (diploid-based) or graph-based coalescent approach (haplotype-resolved). Supplementary Table 9. Summary of super pangenome gene families identified in 144 grapevine genomes. The numbers represent the count of gene members for each gene family. Supplementary Table 10. Summary of NLR gene families identified in 144 grapevine genomes. The numbers represent the count of NLR gene members for each gene family. Supplementary Table 11. The number of genotyped SV. Supplementary Table 12. Summary of expression SV–QTLs significantly associated with downy mildew resistance of Vitis accessions. The simple linear regression model of the MatrixEQTL package (v2.3) was used to detect associations between SV–gene pairs. The Benjamini–Hochberg procedure was applied to control the false discovery rate. Supplementary Table 13. Summary of expression SNP–QTLs significantly associated with downy mildew resistance of Vitis accessions. The simple linear regression model of the MatrixEQTL package (v2.3) was used to detect associations between SNP–gene pairs. The Benjamini–Hochberg procedure was applied to control the false discovery rate. Supplementary Table 14. Summary of transcriptome datasets used for genome annotation in this study. Supplementary Table 15. The oligo sequences used in this study. Supplementary Table 16. The accessions of genome assemblies in NGDC and INSDC.
Source data
Source Data Fig. 6
Unprocessed gels for Fig. 6g.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guo, L., Wang, X., Ayhan, D.H. et al. Super pangenome of Vitis empowers identification of downy mildew resistance genes for grapevine improvement. Nat Genet 57, 741–753 (2025). https://doi.org/10.1038/s41588-025-02111-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-025-02111-7
