
Abstract
The evolution of isocitrate dehydrogenase (IDH)-wildtype glioblastoma (GBM) after standard-of-care therapy remains poorly understood. Here we analyzed matched primary and recurrent GBMs from 59 patients using single-nucleus RNA sequencing and bulk DNA sequencing, assessing the longitudinal evolution of the GBM ecosystem across layers of cellular and molecular heterogeneity. The most consistent change was a lower malignant cell fraction at recurrence and a reciprocal increase in glial and neuronal cell types in the tumor microenvironment (TME). The predominant malignant cell state differed between most matched pairs, but no states were exclusive or highly enriched in either time point, nor was there a consistent longitudinal trajectory across the cohort. Nevertheless, specific trajectories were enriched in subsets of patients. Changes in malignant state abundances mirrored changes in TME composition and baseline profiles, reflecting the co-evolution of the GBM ecosystem. Our study provides a blueprint of GBM’s diverse longitudinal trajectories and highlights the treatment and TME modifiers that shape them.
Similar content being viewed by others


Main
Despite significant advances in basic and translational research, isocitrate dehydrogenase (IDH)-wildtype glioblastoma (GBM)1 remains incurable. The standard treatment for newly diagnosed GBM consists of maximal surgical resection followed by radiotherapy and concurrent treatment with the alkylating agent temozolomide (TMZ). This treatment often elicits a partial response, particularly when the promoter of the O6-methylguanine-DNA methyltransferase (MGMT) gene is methylated, as this gene encodes a DNA-repair protein that can remove alkyl groups added to O6-methylguanine by TMZ. However, the disease invariably recurs with a median overall survival of 14.6 months2,3. To interrogate mechanisms of recurrence, several studies have analyzed the longitudinal evolution of GBM specimens4,5,6. The Glioma Longitudinal Analysis (GLASS) consortium reported high similarity in genetic driver alterations between primary and recurrent GBM, indicating that standard treatment does not exert substantial clonal selection7. Conversely, transcriptome and proteome studies have demonstrated more dynamic evolutionary patterns, such as a proneural-to-mesenchymal (MES) shift, enhanced neuronal signaling and upregulation of extracellular matrix (ECM)-related genes at recurrence5,6,8,9,10,11,12,13,14. However, published longitudinal GBM studies have typically relied on bulk tissue profiling (averaging the signal across the malignant and tumor microenvironment (TME) compartments) and lack the granularity to independently interrogate the evolution of malignant and TME cell types and cell states. Single-cell longitudinal studies have had limited cohort size and limited clinical metadata, precluding a direct analysis of single-cell profiles with clinical correlates14. These limitations, along with the challenge of reconciling results from different groups that leverage diverse analytical and classification schemes, underscore the need for a larger single-cell longitudinal GBM dataset with detailed clinical and genomic annotations and with robust analysis.
We sought to address these challenges in two studies that we present here. We established the GBM Cellular Analysis of Resistance and Evolution (CARE) consortium to characterize the longitudinal evolutionary trajectories in GBM at single-cell resolution. We performed single-nuclei RNA sequencing (snRNA-seq) of paired primary and recurrent GBM samples from 59 patients with detailed clinical annotations and genetic profiles. In the companion study of ref. 15, we conducted an analysis of the data independent of sampling time (primary versus recurrent) that characterized GBM transcriptional ecosystems across multiple layers. In this work, we examine the longitudinal changes in these ecosystems between primary and recurrence and identify molecular and clinical features that modify their trajectories. We show that a lower malignant cell fraction and a reciprocal increase in proportions of glial and neuronal (that is, glio-neural) TME cell types is the most consistent longitudinal change at recurrence, observed in 66% of cases. Contrary to prior studies, we observed no consistent malignant GBM state enrichment at recurrence across the cohort. However, specific trajectories were associated with defined subsets of cases, such as GBMs with MGMT methylation or radiation-related small deletion phenotype. Notably, changes in malignant state abundance were mirrored by corresponding changes in baseline profiles and TME composition, reflecting interdependent alterations of the GBM ecosystem. Our study highlights the diverse longitudinal GBM trajectories that are shaped by treatment response and TME interactions.
Results
Longitudinal profiling of a clinically annotated GBM cohort
We collected matched snap-frozen GBM specimens from primary and recurrent surgeries (Fig. 1a and Supplementary Table 1). Tumor samples were obtained from seven hospitals, resulting in a total of 121 tumors from 59 patients profiled by snRNA-seq and whole-exome sequencing (WES) or whole-genome sequencing (WGS; Fig. 1b; Methods). For the majority of patients, we profiled samples from two time points (labeled T1 and T2); for three patients, we profiled samples from three time points (T1, T2 and T3). Most matched sample pairs were derived from primary and first recurrent tumors, except for three pairs that consisted of two recurrent samples and four pairs that consisted of a primary and a second recurrence.

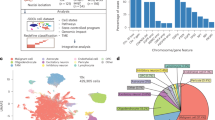
a, Schematic representation describing the workflow of our study. TMZ, temozolomide; RT, radiotherapy; int., intermediate. b, Clinical and technical information on all 59 patients in this dataset, including surgical interval, age at diagnosis, gender, ___location at recurrence (rec), MGMT promoter status, therapy and omic data type. c, UMAP for dimension reduction based on gene expression values of the cells in our cohort colored according to the cell type (top) and time point (bottom). d, Proportion (%) of patients in the dataset in which a genetic aberration was detected, colored by time point. The donut chart at the top represents average somatic mutation distribution for whether an alteration was detected at both time points (T1 + T2), the primary tumor only (T1) or the recurrent tumor only (T2). The bar plot at the bottom is for the known driver genomic events and the representative phenotypes related to the genomic alterations—arm-level refers to whole chromosome arm amplification (amp) (chr7q)/deletion (del) (chr10q), CNA refers to high-level gene amplification or deletion, and SNV refers to single-nucleotide mutations. De novo hypermutation (HM) indicates a high mutation burden at both time points without evidence for treatment-associated mutational signatures. Small deletion indicates tumors with increased small deletion burden at recurrence. UMAP, uniform manifold approximation and projection; chr, chromosome; assoc., associated.
Tumor ___location was known for 51 patients, of whom 45 cases locally recurred at the surgical margin and six cases recurred at a site distant from the resection cavity. The majority of patients (51/59) received radiotherapy with concurrent TMZ and adjuvant TMZ following surgery. The improved clinical outcomes in our cohort (median interval between two surgeries = 12.4 months, median overall survival = 24 months), compared with historical cohorts, likely reflect better performance status of patients eligible for multiple surgical resections, consistent with a younger age at diagnosis (median = 53 years, range = 19–73 years).
In our 10x Genomics snRNA-seq dataset, 429,305 cells passed quality filters (Methods). Cells were classified as malignant (246,408 cells) or nonmalignant (182,897 cells) based on inferred chromosomal copy-number aberrations (CNA). Nonmalignant cells were further classified and annotated based on established cell-type signatures and marker gene expression16,17,18,19 (Fig. 1c; Methods). Nonmalignant cells clustered together across time points (Fig. 1c, bottom), and we did not observe a significant difference in the number of cells profiled or the number of genes detected per cell between time points (Extended Data Fig. 1a,b).
Mutational and CNA profiles were derived from bulk DNA-sequencing data (Fig. 1b). The frequency of GBM driver alterations was consistent with prior studies7 and remained largely stable across time points (Fig. 1d and Extended Data Fig. 1c). However, 26% of cases (14 of 54 patients) acquired treatment-related DNA hypermutation20 or an increase in small deletion burden21 at recurrence (Fig. 1d, right).
Longitudinal changes in cell-type composition
Global (nonmatched) comparison of cell-type abundance showed overall similarity between time points, except for significantly lower abundance of malignant cells and higher abundance of glio-neural TME (oligodendrocytes, neurons and astrocytes (ACs)) in recurrent tumors (Fig. 2a and Extended Data Fig. 2a). We further separated the broad nonmalignant cell types (for example, myeloid) into more granular states (for example, microglia and bone marrow-derived macrophages) using non-negative matrix factorization (NMF; Methods). Analysis of these higher-resolution cell states demonstrated that recurrences were enriched with reactive ACs and with oligodendrocytes expressing an interferon program (Extended Data Fig. 2b). To further evaluate differences in cell-type composition, we categorized tumors into six compositional groups based on the relative abundance of malignant and TME cell types (Methods). Briefly, tumors were divided into the following three main groups based on their malignant fraction: high malignant fraction (HMF), intermediate malignant fraction (IMF) and low malignant fraction (LMF). The latter group was further subdivided by the dominant TME cell type (for example, LMF—tumor-associated macrophages (TAM), oligodendrocyte (OC) and glio-neural (GN); Extended Data Fig. 2c, left). The HMF group was enriched in primary tumors, while the LMF with glio-neural (LMF—GN) or oligodendrocytic TME (LMF—OC) were enriched at recurrence (P = 0.005 for both groups, two-sided Fisher’s test; Fig. 2b, left).

a, Proportion (%) of each cell type across the two time points (primary and recurrence, n = 118) in which a statistically significant difference was observed. P values were computed with a two-sided Wilcoxon rank-sum test. b, Group-differentiating features and breakdown across primary/recurrence status for composition (left), malignant state frequency (middle) and baseline profiles (right). P values were computed using Fisher’s exact test. c, Kaplan–Meier curves depicting the survival time after the second resection according to the transcriptomic features at the second resection. Left, 12 patients that recurred with a glio-neural composition group versus 47 patients with other types of recurrences. Right, six patients that recurred with MES/hypoxia cell state group versus 53 patients with other types of recurrences. X axis reflects the time from the second resection to outcome. Statistical significance of survival difference between the groups in each panel was computed using the log-rank test. d, Heatmap showing the Pearson correlation between transcriptomic features. Correlations were computed separately in primary samples (top-left) and in recurrent samples (bottom-right) and are ordered according to the hierarchical clustering pattern of the recurrent samples. NS, not significant.
To validate our findings, we re-analyzed an independent longitudinal GBM snRNA-seq cohort14 (Extended Data Fig. 2d,e). Consistent with our cohort, we observed lower malignant cell abundance, higher oligodendrocyte abundance and similar enrichment of LMF compositional groups at recurrence (P = 0.04, two-sided Fisher’s exact test; Extended Data Fig. 2f).
Survival analysis stratified by compositional groups showed significant differences in survival time after the second surgery (Extended Data Fig. 2g, top) but not in the time interval between the first and second surgeries. Patients with glio-neural TME at recurrence had significantly longer survival time after second surgery than patients with other recurrences (median = 19 versus 8 months, P = 0.012, log-rank test; Fig. 2c, left). These differences in clinical course were independent of other clinical characteristics, including MGMT methylation status (Extended Data Fig. 2h, top).
Longitudinal stability of transcriptional features
Next, we investigated changes in malignant cell state abundance across time points. As described in more detail in the companion paper15, we applied NMF to malignant cells and identified ten GBM metaprograms (MPs) representing intratumoral cellular heterogeneity (Methods)22,23,24. This included the cell cycle and the previously identified AC-like, MES-like, stress, hypoxia, neural progenitor cell-like (NPC-like) and oligodendrocyte progenitor cell-like (OPC-like) states. In addition, we identified the following three previously undescribed states: a differentiated neuronal state (NEU-like), a glial progenitor-like state (GPC-like) and a cilia-related state (Cilia-like), characterized by marker genes, pathway enrichments and similarity with normal neural cell types. Beyond cell states, we defined three baseline gene expression profiles (BP), reflecting intertumor heterogeneity that is not driven by differences in malignant cell state abundance. The three BPs were annotated as glial-BP, ECM-BP and neuronal-BP.
Comparison of malignant cell state abundance between primary and recurrent samples did not reveal significant differences both in our cohort and in the cohort of ref. 14 (Extended Data Fig. 3a,b). Moreover, we did not detect significant differences across time points when considering a division of tumors into six groups based on dominant malignant states or by BPs (Fig. 2b (middle and right), Extended Data Fig. 2c (middle and right) and Extended Data Fig. 3c,d). However, survival analysis stratified by dominant malignant cell states showed significantly worse prognosis in the MES-like/hypoxia group at recurrence (median survival time from second surgery = 5 versus 10.3 months, P = 0.00086, log-rank test; Fig. 2c (right) and Extended Data Fig. 2g,h), consistent with a recent study14.
The multiple transcriptomic layers of cell-type composition, malignant cell states and malignant BPs are significantly associated with each other and define three stereotypical GBM ecosystems (that is, glial, neuronal and ECM)15. We next investigated whether interactions between components of the GBM ecosystems are maintained between primary and recurrent tumors. We separately computed for each time point the correlations between (1) proportions of 11 TME cell types, (2) proportions of ten malignant cell states and (3) three BP scores. In both time points, we found three comparable clusters of correlated features (Fig. 2d and Extended Data Fig. 3e). These conserved associations (P < 0.01 by permutation test) remain largely stable.
Nevertheless, we did observe some differences, such as preferential association of the neuronal-high ecosystem with OPC-like malignant cells (in primary samples) and with NEU-like malignant cells (in recurrent samples; Fig. 2d and Extended Data Fig. 3e). Interestingly, ligand–receptor analysis with CellChat25 showed some differential enrichment of predicted interactions across time points. For example, neuroligin (NLGN) and neuregulin (NRG) gene families—expressed by oligodendrocytes and neurons—are predicted to interact with the neurexin (NRXN) gene family or ERBB4 on NEU-like and NPC-like malignant cell states, preferentially at recurrence (Extended Data Fig. 3f)15,26,27,28. Taken together, global analysis of our cohort (not matched per sample pairs) shows differences in cell-type composition that are consistent with decreased malignant cell fraction and further integration of recurrent GBM cells with the brain parenchyma9,29,30, but does not reveal substantial global differences either in malignant cellular states or in BPs.
Divergence of individual sample pairs
Despite global conservation of cell types and malignant cellular states, when examining individual matched sample pairs, we found extensive divergence (Fig. 3a). Most (68%) sample pairs diverged from one compositional group at diagnosis to a different one at recurrence. Similarly, 64% and 56% switched groups of dominant malignant states and of BP, respectively. We observed the same phenomenon in the cohort of ref. 14, with 68% of patients switching compositional groups and 72% switching malignant groups at recurrence (Extended Data Fig. 4a). While no group was significantly enriched, there was a trend toward increased neuronal-BP and reduced glial-BP at recurrence (Fig. 3a (right)). Remarkably, we observed 72% of all possible trajectories at least once (63 of 88 trajectories), including opposite trajectories. For example, patient P1 had an increase of AC-like and decrease of GPC-like malignant cells at recurrence, while patient P2 had an opposite trajectory (Extended Data Fig. 4b). Furthermore, patients profiled at three time points showed opposite trajectories between two treatment intervals (T1–T2 and T2–T3), despite recurrences occurring at the same site (Fig. 3b). Finally, the frequency of conservation was not significantly different between local and distant recurrences (P = 0.66, two-sided Fisher’s test).

a, Sankey plots depicting the transitions between time points across the composition (left), malignant states (middle) and baseline profile groups (right). The width of the features represents the proportion of tumors with the indicated classification. Light gray links reflect transitions from one group (T1) to a different group at T2. b, Longitudinal transcriptomic changes in the two pairs profiled at three time points highlight unpredictable gene expression trajectories. Detailed clinical information including treatment regimen and tumor MRI (top), heatmap representing proportions and scores from three transcriptomic layers (middle) and mutation fraction between time points are shown (bottom). INF, interferon; BEV, bevacizumab; T1Gd, T1-weighted post-gadolinium image. c, Observed (colored bar) versus expected (black dot) conservation across time points. P values were computed using a binomial test and adjusted for multiple testing using Holm’s method. Error bars represent confidence intervals derived from the binomial test. Asterisk indicates a Holm-adjusted P < 0.01. MRI, magnetic resonance imaging.
This divergence of individual sample pairs prompted us to compare the observed patterns of conservation to those expected by a null model in which matched primary–recurrent pairs are as similar to one another as pairs of randomly selected samples. This analysis revealed that, across all three layers, matched pairs were significantly more conserved than expected by chance (P < 0.004 for each layer, binomial test; Fig. 3c). Thus, matched samples usually diverge in their transcriptomic classification and may diverge in a wide range of trajectories between and within patients but remain significantly more likely to retain any specific classification compared to a random pair of samples. This observation prompted us to further stratify our cohorts and attempt to identify predictors/modifiers of evolutionary trajectories.
MGMT expression in GBM single cells influences recurrence trajectories
Previous studies have shown that patients with methylated MGMT promoter exhibit better response to TMZ and have more favorable prognosis2,31, but whether this is mirrored by differences in either cellular architecture or in cellular trajectories following treatment is unknown. We classified patients as methylated (MGMT-MET, n = 12), unmethylated (MGMT-UM, n = 19) or unknown (n = 28), based on the primary tumor’s MGMT promoter methylation status. As expected, MGMT-MET patients exhibited longer surgical intervals than MGMT-UM patients, both in our cohort and in the GLASS cohort (Extended Data Fig. 5a,b).
We next assessed whether MGMT promoter methylation status correlated with MGMT expression in single cells, as this association has been challenging to establish in bulk analyses32. Malignant cell expression of MGMT was significantly lower in primary MGMT-MET samples than in primary MGMT-UM samples (P = 5.3 × 10−6, two-sided Wilcoxon rank-sum test; Fig. 4a)32,33, but this effect was not observed for TME cell types (Fig. 4b). In a subset of MGMT-MET cases, MGMT expression increased at recurrence, possibly reflecting a competitive disadvantage of cells with low MGMT expression during treatment.

a, Average MGMT expression in malignant cells per sample across time points. Samples were included only if the MGMT promoter status was verified using a methylation assay and were annotated as methylated/unmethylated according to the status of the primary sample (n = 12 methylated and n = 19 unmethylated). Dashed lines connect matched samples. Statistical significance was estimated using a two-sided Wilcoxon rank-sum test; unadjusted P values are shown. In a, for within-group comparisons, a Wilcoxon signed rank test (paired) was used. b, MGMT expression level in each cell type between methylated and unmethylated samples. Statistical significance was estimated using a two-sided Wilcoxon rank-sum test; unadjusted P values are shown. c, Survival curve for the patients included in this analysis stratified according to MGMT expression level. X axis reflects the time from first to second resection. Statistical significance of survival difference between the groups in each panel was computed using the log-rank test. d, Survival curves for MGMT-MET patients stratified according to MGMT expression level. X axis reflects the time from first to second resection. Statistical significance of survival difference between the groups in each panel was computed using the log-rank test. e, Survival curves for MGMT expression high group stratified according to MGMT promoter methylation status. X axis reflects the time from first to second resection. Statistical significance of survival difference between the groups in each panel was computed using the log-rank test. NOS, not otherwise specified. f, Difference in MES-like cell state proportion (%) across the two time points (primary and recurrence) stratified by MGMT expression level in this study and in ref. 14. All sample points are shown as jittered points. Statistical significance was estimated using a two-sided Wilcoxon rank-sum test; unadjusted P values are shown. g, MGMT-specific signatures plotted for patients included in the analysis. Value represents the T2–T1 difference (log2(FC) of gene expression) for each patient. Patients are ordered according to the difference between signature scores. Genes are ordered according to their correlation (across patients) with the difference between signature scores. FC, fold change.
The correlation between MGMT promoter methylation and low MGMT expression prompted us to build a logistic regression model that uses average malignant cell MGMT expression to infer MGMT methylation status (Extended Data Fig. 5c). When applying this model, we found that classification by MGMT expression levels was associated with surgical resection interval (Fig. 4c) and, in fact, led to improved association compared to classification by MGMT promoter methylation alone—within the MGMT-MET group, we observed significantly longer surgical interval for the MGMT-low expression group relative to the MGMT-high/MGMT-intermediate expression groups (Fig. 4d); in contrast, within the MGMT-high expression group, there was no significant difference in surgical interval when further stratifying by MGMT promoter methylation status (Fig. 4e). This improved performance of MGMT expression level could be due to exclusion of nonmalignant cells that may affect MGMT promoter methylation assessment; alternatively, it could indicate that MGMT expression level in the therapeutically targeted malignant cells more directly reflects MGMT protein activity and hence is more predictive of treatment response.
Comparison of cellular architecture between MGMT-low and MGMT-high expression samples showed no significant differences in cell-type composition and malignant state distribution (Extended Data Fig. 5d,e). However, MGMT expression level was significantly associated with changes in abundance of MES-like states at recurrence, both in our CARE dataset and in ref. 14 (Fig. 4f and Extended Data Fig. 5f). Specifically, MGMT-low tumors recurred with a lower abundance of MES-like states compared to their primary samples, while MGMT-high tumors displayed the opposite pattern. Baseline profiles changed in a similar manner—trending toward decreased ECM in MGMT-low, highlighting the coordinated evolution of cellular states and BPs (Extended Data Fig. 5g).
To further identify longitudinal gene expression patterns specific to MGMT-high and MGMT-low samples, we compared their malignant pseudobulk expression (aggregated gene expression from all malignant cells per sample) across time points. This revealed four groups of genes—those consistently upregulated or downregulated in recurrence, regardless of MGMT expression (CONS-UP and CONS-DN), and those upregulated specifically in MGMT-low (low-UP) or MGMT-high samples (high-UP; Extended Data Fig. 5h). CONS-UP was enriched with genes involved in postsynaptic activity (for example, GRIN1/GRIN2A/GRIN2B, GABRA1/GABRA4), consistent with the trend toward increased neuronal-BP in recurrence (Fig. 3a), while CONS-DN was enriched with genes related to oxidative phosphorylation (for example, NDUFB4/NDUFB11, UQCRB/UQCRQ) and stemness (for example, CD24, DCX, SOX4/SOX11 and SOX11; Extended Data Fig. 5i). In contrast to those MGMT-independent gene sets, low-UP genes were enriched with genes linked to gliogenesis (for example, ACSL1, OLIG1/OLIG2 and SOX8), whereas high-UP genes were enriched with mesenchyme development and ECM remodeling (for example, COL1A1/COL1A2, COL1A2, DCN, FN1 and LOXL1; Fig. 4g). To validate our findings, we repeated this analysis in the ref. 14 dataset and found significant similarities, especially in the high-UP signature (Jaccard index 8.5% versus 1.2% expected, P < 0.0001, bootstrapping).
In summary, our single-cell analysis revealed that MGMT expression in malignant cells predicts time to recurrence and influences longitudinal cellular trajectories—MES-like state and ECM-related genes tend to be enriched in recurrence in MGMT-high (likely nonresponsive) patients, while in MGMT-low (likely responsive) patients, recurrence is associated with loss of MES-like states and enrichment of genes related to gliogenesis. Our results suggest that prior attempts that failed at correlating MGMT expression with treatment response32 were likely confounded by bulk analysis and expression of MGMT by nonmalignant cells.
Mutational signatures associated with recurrence trajectories
Treatment may affect GBM genetic evolution, as supported by reports of somatic hypermutation following TMZ chemotherapy and/or small deletion phenotype following radiotherapy7,20,21,34. To analyze the longitudinal mutation patterns in the CARE cohort, we examined mutation burden and mutational signatures following therapy. We observed that mutation burden (mutations per megabase) generally increased at recurrence (P = 0.01, two-sided paired Wilcoxon; Extended Data Fig. 6a) and identified four treatment-associated hypermutant samples (mutation burden > 10 mutations per megabase (Mut per Mb) at recurrence; Methods)34. Two tumor pairs displayed high mutation burden at both time points without evidence of treatment-associated mutation signature (COSMIC single base substitution (SBS) 11; Extended Data Fig. 6b).
To investigate new mutational signature associations at recurrence, we examined the frequency of the COSMIC v3 SBS signatures35, both in our CARE cohort (n = 46 patients) and in the GLASS cohort (n = 138 patients). In both cohorts, we observed a significant decrease in SBS1 (aging/clock-like etiology) and an increase in SBS21 (defective mismatch repair) at recurrence (Fig. 5a and Extended Data Fig. 6c). Notably, patients with increased mutations attributed to SBS21 did not tend to acquire mutations in mismatch repair genes. While mutational signatures temporally differed, we did not observe individual mutations enriched at recurrence (adjusted Fisher’s exact test P > 0.05). Among small insertion/deletion variants, we observed increased burden of deletions (Fig. 5b) but not insertions (Extended Data Fig. 5d), including ten recurrences that acquired the small deletion phenotype without treatment-associated hypermutation (Methods; Fig. 5b). Taken together, our analyses reveal that subsets of tumors acquire consistent patterns of genetic alterations following treatment.

a, Longitudinal difference in the relative proportion of mutations assigned to COSMIC v3 mutational signatures at each time point for select SBS signatures (SBS1, clock-like; SBS11, alkylating agent; and SBS21, defective DNA mismatch repair). Outlier points are shown. Dashed lines connect values from the same patient at T1 and T2. P values represent two-sided paired Wilcoxon rank-sum test. b, Longitudinal difference in small deletion burden between T1 and T2 with dashed lines connecting patient values and red indicating patients who were assigned to the increased small deletion burden group. Analysis restricted to nonhypermutant cases. P values reflect two-sided paired Wilcoxon rank-sum test. c, A waterfall plot of the relative change in hypoxia-related malignant state sorted by decreasing longitudinal change. Patients are represented by a single bar (x axis). Presence of increased small deletion burden or SBS21 signature is presented in black annotation bars below. The reported P values indicate two-sided paired Wilcoxon rank-sum test for the longitudinal change in the hypoxia malignant state for those patients with small deletion or SBS21 increase. d, Bulk RNA data from the GLASS consortium were scored for the hypoxia snRNA malignant metaprogram signature and the relative ssGSEA score is presented on the y axis. The panels show the longitudinal change in the hypoxia signature for those patients whose tumor had a small deletion increase (left) and for those that did not have small deletion burden increase (right). P values from two-sided paired Wilcoxon rank-sum test. e, Scatterplot that represents the scaled genetic distance (bulk DNA—proportions of private mutations and differences in copy-number altered regions) versus scaled transcriptional distance (snRNA), with the linear regression line shown in blue. The Pearson’s correlation coefficient and associated P value are shown (n = 38). ssGSEA, single-sample gene set enrichment analysis; diff., differences.
To understand whether post-treatment genetic alterations modify cellular state evolution, we investigated the specific trajectories of tumors with hypermutation, SBS21, and small deletion phenotypes. For hypermutant tumors, we did not observe a consistent cellular phenotype at recurrence, although this analysis was limited by sample size (n = 4). In contrast, patients who acquired small deletions were enriched with hypoxia and MES-like cell states (Fig. 5c and Extended Data Fig. 6e; paired Wilcoxon rank-sum test, P < 0.05). We observed a similar effect for tumors with increased SBS21 signal, although this effect was weaker when excluding samples with co-occurring small deletion phenotype (Fig. 5c and Extended Data Fig. 6f). The association between recurrence-specific small deletions and hypoxia state was recapitulated in analysis of bulk samples from GLASS9 (Fig. 5d). In contrast, the MES-like observation did not validate in the GLASS cohort (Extended Data Fig. 6g), possibly due to similarities between MES-like and nonmalignant profiles that make it difficult to disentangle these signals in bulk datasets. Hypoxic cells have been linked to deregulated epigenetics and stress responses36 and, consequently, to increased resistance to radiation37. Thus, malignant cells in hypoxic regions may be better able to repair radiotherapy-associated DNA damage, leading to an outgrowth of hypoxic state at recurrence.
Finally, we examined the correlation between overall genetic distance, as defined by longitudinal single-nucleotide variant (SNV)/CNA differences, and overall malignant transcriptional distance (Methods). We found a significant correlation (Pearson’s correlation = 0.42, P = 0.009; Fig. 5e) that was retained when analyzing only SNV or CNAs (Extended Data Fig. 6h,i), suggesting that intratumoral genetic evolution during disease progression contributes to transcriptional evolution.
Discussion
Intertumoral and intratumoral heterogeneity in GBM has been associated with treatment resistance and disease recurrence, but how specific subsets of GBM cells respond to therapy remains poorly understood. Here we leveraged a longitudinally matched multi-omic GBM snRNA-seq and bulk DNA dataset of 121 tumors from 59 patients consisting of ~430,000 nuclei to comprehensively characterize the compositional, transcriptional and genetic changes that GBMs undergo following standard-of-care. We found extensive variability in longitudinal trajectories of different patients (Fig. 6). While many trajectories were unpredictable, we identified patient characteristics—MGMT methylation and increased small deletion burden—that were associated with particular trajectories and expanded our understanding of recurrences.

Analysis of the GBM CARE dataset reveals key patterns of GBM evolution following treatment. (1) The most common observation was in the cell-type composition layer, where many samples exhibited a reduced malignant cell fraction at recurrence. (2) At the malignant state level, many tumors recurred with an unpredictable trajectory that likely reflects patient-specific evolution (top). A smaller subset of tumors recurred with more predictable trajectories, such as reduced MES-like fraction for MGMT-MET versus MGMT-UM primary tumors (middle) or increased hypoxia fraction for samples with increased small deletion burden following treatment (bottom). Columns separated into malignant and nonmalignant fractions indicate the relative proportions per tumor. Only selected nonmalignant and malignant cell states are shown for clarity. Hyp, hypoxia.
Previous studies have highlighted specific transcriptional trajectories during GBM evolution. The recent GLASS consortium study highlighted three trajectories toward neuronal, MES and proliferative profiles9, and other studies have highlighted proneural-to-MES phenotypic shifts11,14,38. While we similarly identified these recurrence-associated patterns, our analysis suggests that their overall frequency is limited and that such trajectories are enriched only in specific subsets of patients, while global comparison of cell states between primary and recurrent samples shows overall similarity. In particular, a shift toward the MES phenotype following treatment was not observed as a globally enriched trend in our dataset, only in specific subsets of patients. This discordance with prior studies may be attributable to at least the following three effects: (1) differences in technology (for example, bulk versus single-cell profiling), (2) cohort/sampling (for example, single-center sampling, tissue digestion versus not) and (3) analysis methods. There may also be biases due to additional treatments that were not addressed (bevacizumab, corticosteroids, etc.).
Our dataset contains tumor samples that were collected from seven medical centers across six countries, limiting the possibility of results being skewed toward a particular recurrence trajectory due to institution-specific procedures. Moreover, the samples in our dataset were processed and profiled by three different labs, limiting the possibility of skewing due to a lab-specific batch effect. The snRNA-seq protocol used by our labs to process and profile tumor samples was digestion-independent and optimized for brain tumors39, capturing a higher number of transcripts than prior single-nucleus GBM studies (Extended Data Fig. 2e). This likely results in a more faithful representation of the tumor tissue, as evident by the higher diversity of TME cell types in our dataset and the discovery of new malignant cell states.
Neuronal pathways were consistently found to be upregulated in recurrent GBM8,9,12. However, it is important to note that this reflects only subsets of patients in each study and that we observe a wide diversity of trajectories from primary to matched recurrent samples. Leveraging the resolution and scale of our dataset, we showed that this phenomenon correlates with decreased malignant cell fraction and concomitant increased proportions of neurons in the TME; only in a small subset of cases, this is accompanied by the enrichment of NEU-like malignant state and the neuronal baseline expression profile of the tumors. These results suggest that, following treatment, integration of the residual GBM cells with the brain parenchyma is likely enhanced29,30.
Beyond defined subsets, our study makes the challenging observation that, for the majority of GBMs, longitudinal cellular trajectories are diverse and unpredictable. We speculate that this diversity reflects at least the following three phenomena: first, given the genetic and epigenetic heterogeneity in primary tumors, it might be expected that tumors with distinct underlying biology will also have distinct trajectories of recurrence. Second, even within a single GBM, there is extensive spatial heterogeneity23,40; as primary and recurrent samples are obtained from different locations, their differences might reflect such spatial heterogeneity and, therefore, depend on sampling40. Third, patients vary considerably both in their treatment histories and in their response to treatments, potentially leading to distinct recurrence trajectories. Many GBM patients are treated by additional therapies in diverse combinations beyond standard-of-care, which is a limitation in this study. Moreover, even standard-of-care induces distinct patient responses, and we identified the following two cases in which treatment responses are linked to recurrence trajectory: (1) MGMT-MET (MGMT-low expression) samples (more likely to respond to alkylating agents) are associated with depletion of MES-like states in recurrence and (2) the small deletion phenotype, previously linked to radiotherapy21, is associated with increased hypoxia states in recurrence.
In summary, we present a comprehensive analysis of GBM recurrence at single-cell resolution. Our analysis shows that, globally, recurrent GBMs are generally similar to primary GBMs in their cellular states, composition and other features of the GBM ecosystem. Accordingly, a wide range of recurrence trajectories are observed, highlighting extensive diversity in GBM evolution. Nevertheless, subsets of tumors with evidence for particular treatment responses are skewed toward specific transcriptional trajectories. These findings offer more detailed insights over prior observations and provide an opportunity to stratify recurrent patients more precisely, potentially leading to specific treatment strategies.
Methods
Experimental method details
Human participants and ethical approval
Frozen GBM specimens that were diagnosed as ‘glioblastoma, IDH-wildtype’ according to the World Health Organization 2021 classification were collected at seven institutions, listed below. Collection and genomic profiling were approved by the institutional review board of each institution, and all patients provided informed consent accordingly. The tumors were collected from the MD Anderson Cancer Center, approved by the Institutional Review Board of MD Anderson Cancer Center under the protocol 2012-0441. Duke University cohort (n = 12)—the tumors were collected from the Preston Robert Tisch Brain Tumor Center Biorepository, Duke University Hospital. It was approved by the Institutional Review Board of Duke University under the protocol Pro0007434. Tokyo University cohort (n = 30)—the tumors were collected from the Department of Neurosurgery, Tokyo University Hospital. It was approved by the Institutional Review Board of Tokyo University Hospital under the protocol G10028. Pitié-Salpêtrière Hospital cohort (n = 28)—the tumors were collected from Pitié-Salpêtrière Hospital. It was approved by the Onconeurotek Brain Tumor Bank of the hospital Pitié-Salpêtrière certification 96-900. St. Michael’s Hospital cohort (n = 14)—the tumors were collected from the Division of Neurosurgery, St. Michael’s Hospital, Unity Health Toronto. It was approved by the Research Ethics Board of St. Michael’s Hospital, Unity Health Toronto, under the protocol REB 13-141, and all patients provided signed informed consent accordingly. Seoul National University (SNU) cohort (n = 20)—the tumors were collected from the Department of Neurosurgery, SNU Hospital. It was approved by the Institutional Review Board of SNU Hospital (approval H-2004-049-1116). NORLUX Neuro-oncology Laboratory (NORLUX) cohort (n = 13)—the tumors were collected from Centre Hospitalier de Luxembourg (CHL, Neurosurgical Department). It was approved by the National Committee for Ethics in Research, Luxembourg, under the protocol 201201/06. Cohorts were added to the Institutional Review Board protocol Dana–Farber/Harvard Cancer Center 10-417. Patients were male and female. Clinical information of the cohorts is summarized in Supplementary Table 1.
Statistics and reproducibility
No statistical method was used to determine the sample size. Data were excluded from select analyses when the number of malignant cells within a sample was low in at least one time point. Throughout the figures in this manuscript (Figs. 2–5 and Extended Data Figs. 1–3, 5 and 6), the boxplots reflect the following summary statistics: (1) the line splitting the box represents the median value; (2) the lower and upper edges correspond to the first and third quartiles (the 25th and 75th percentiles); (3) the upper whisker extends from the edge to the largest value no further than 1.5× interquartile range (IQR) from the edge; (4) the lower whisker extends from the edge to the smallest value at most 1.5× IQR of the edge; and (5) if the plots do not include jitter points representing the actual data, then data beyond the end of the whiskers are plotted individually as outlying points. The statistical analysis described in this work was done using R version 4.0.1 and above.
Nuclei isolation from frozen tissue
Protocol 1 (laboratory 1 and laboratory 2 for the cohorts from Duke University, MD Anderson Cancer Center, Tokyo University Hospital, St. Michael’s Hospital and Pitié-Salpêtrière Hospital)—nuclei from frozen tumor tissue were isolated as follows: tissue was thawed and mechanically dissociated in salt-Tris buffer (10 mM Tris–HCl (pH 7.5), 146 mM NaCl, 1 mM CaCl2 and 21 mM MgCl2) with 0.49% CHAPS (3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate) (Millipore, 28300). Single-nuclei suspensions were filtered using a 40 μm strainer, centrifuged at 500g for 5 min and resuspended in salt-Tris buffer supplemented with 0.01% BSA (NEB, B9000S). Nuclear suspensions were inspected by microscope, counted using a hemocytometer and used for 10x Genomics workflow.
Protocol 2 (laboratory 3 for the cohorts from NORLUX and SNU cohorts)—tissue samples were thawed and mechanically dissociated in nuclei EZ lysis buffer (Millipore Sigma, NUC101) via Dounce homogenization. The solutions were incubated on ice for 5 min and mixed one to two times during incubation. Single-nuclei suspensions were filtered through a 70 μm strainer and centrifuged at 500g for 5 min at 4 °C, resuspended in nuclei EZ lysis buffer and incubated on ice for 5 min. The solutions were centrifuged at 500g for 5 min at 4 °C and resuspended in 1% BSA/0.2 U μl RNase inhibitor/PBS buffer (three times). For the final resuspension, the DAPI was added to the buffer, the solution was filtered through a 40 μm strainer, cells were counted on a Countess II automated cell counter (Thermo Fisher Scientific) and nuclei were taken into the 10x Genomics workflow.
10x Genomics for single-nucleus sequencing
The 10x Chromium Single Cell 3′ Reagent Kit v3 (10x Genomics, PN1000128) was used according to the manufacturer’s protocol. In brief, nuclei were loaded on the Chromium Chip (10x Genomics, PN12000120) with a target cell recovery of 6,000–8,000 nuclei and processed in the Chromium Controller. Single nuclei were partitioned into gel beads-in-emulsion (GEMs), followed by RNA reverse transcription with barcoding. Libraries were created by breaking GEMs and pooling barcoded fractions, cDNA amplification, fragmentation and attachment of sample index and adapter and sequenced on NextSeq500 or NovaSeq (Illumina). For NORLUX and SNU cohorts, nuclei were loaded on a Chromium chip with a target cell recovery of 6,000 nuclei for single-cell multiome ATAC and gene expression according to the manufacturer’s protocol. The gene expression chemistry for the 10x multiome possesses the same chemistry as the 10x v3 3′ snRNA-seq.
WES
WES for the samples from Duke University, MDACC, Tokyo University Hospital and St. Michael’s Hospital was performed as follows: DNA was extracted from each frozen tumor sample and blood sample corresponding to the patients using the DNeasy Blood & Tissue kit (Qiagen, 69504). The genomic DNA (100–250 ng) was acoustically sheared by an ultrasonicator (Covaris), targeting 150 bp fragments. Library preparation was performed by KAPA HyperPrep Kit (KAPA Biosystems, KK8504) followed by the enzymatic clean-up using AMPure XP beads (Beckman Coulter, A63882). Exome capture was performed using a custom exome bait (manufactured by Twist Biosciences to Broad Institute’s specification). Captured libraries were sequenced with 150-base-pair paired-end sequencing on a NovaSeq 6000 (Illumina). For the samples from Pitié-Salpêtrière Hospital, after the DNA was fragmented by a LE220 ultrasonicator (Covaris) and size selected, library preparation and capture were performed using the Twist Human Core Exome kit (Twist Bioscience) according to the manufacturer’s protocol. Sequencing was performed on a NovaSeq 6000 (Illumina) with 200-base-pair paired-end sequencing.
WGS
Newly generated whole-genome DNA-sequencing data were collected for a cohort of frozen samples from the NORLUX Neuro-oncology Laboratory. Briefly, DNA was extracted from each tumor sample using the AllPrep DNA/RNA Minikit (Qiagen, 80204) for samples with sufficient tumor tissue and matched normal blood when it was available. Note that the selected tissue for bulk DNA isolation was adjacent to the tissue used for single-nuclei data generation. Briefly, DNA was sheared to 400 bp using a LE220 ultrasonicator (Covaris) and size-selected using AMPure XP beads (Beckman Coulter, A63882). Whole-genome libraries were prepared and sequenced with 150-base-pair paired-end sequencing on a NovaSeq 6000 (Illumina). WGS data for the SNU cohort were prepared identically and were previously reported in ref. 9. Data for the SNU cohort are available on Synapse (https://www.synapse.org/glass).
Somatic variant detection and analysis
DNA-sequencing alignment, fingerprinting, somatic variant detection (Mutect2) and copy-number segmentation were performed in accordance with the Genome Analysis Toolkit (GATK) best practices using GATK 4.0.10.1, as previously described7,9. Briefly, whole-exome and whole-genome reads were aligned to the b37 genome (human_g1k_v37_decoy) using BWA MEM 0.7.17. DNA fingerprint analysis using ‘CrosscheckFingerprints’ (Picard) confirmed that all samples belonging to a patient came from the same individual, indicating that there were no sample mismatches in this study. Somatic mutations were detected using Mutect2 (v4.1.0.0) for tumor samples with a matched normal blood sample. A panel of normals was constructed for each sequencing batch to account for differences in DNA library prep (for example, whole genome versus whole exome) and used to filter out common artifactual and germline variants. Patient tumor samples without matched normal blood were analyzed for copy-number alterations and tumor-only SNV detection was performed with a panel of normal reference according to GATK best practices. Tumor-only somatic variants were used to identify hypermutant tumors. To identify samples with increased small deletion mutation burden, the following thresholds were applied: the recurrence-specific small deletion mutation burden needed to be greater than 0.2 Mut per Mb sequenced and greater than 0.1 Mut per Mb increase when comparing all small deletion variants in the recurrence versus the primary tumor. Mutational signature estimation was performed using COSMIC v3 signatures for the GLASS and CARE datasets according to previously published methods41. Briefly, all WGS samples were used to generate a mutational matrix and extract de novo signatures using SigProfilerExtractor (v.1.1.4)41 using maximum signatures set to 10 and NMF replicates set to 100. The de novo signatures were then deconvoluted to COSMIC v3.3 signatures. Finally, mutations for each sample were assigned to the most probable signature among the deconvoluted COSMIC signatures using the R package Palimpsest (v.2.0.0)42. Acquired SBS21 samples were denoted as those that were in the top quartile of SBS21 change at T2. For samples with matched blood, hypermutation was determined according to the cutoff of 10 Mut per Mb and further separated into de novo versus treatment-associated based on COSMIC mutational signatures. Treatment-associated hypermutation in samples with matched normal blood was classified by a longitudinal increase in mutation burden (that is, from <10 Mut per Mb to >10 Mut per Mb) and SBS11 signal (alkylating agent-associated signature). For samples without matched blood, a single recurrent sample with a large increase in mutation burden at recurrence compared to the initial tumor (18.3-fold increase) and SBS11 signature was indicative of treatment-associated hypermutation. Genetic distance between time-separated samples from the same patient was defined as the bulk DNA proportion of private mutations (SNV) and differences in CNA segments between the two samples.
Single-cell/single-nucleus data processing of human glioma samples
For droplet-based snRNA-seq data, the Cellranger v3.1.0 pipeline provided by 10x Genomics was used for alignment (GRCh38 release 93) and to generate count matrices. We quantified the gene expression levels as \(\scriptstyle{E}_{i,j}=\log_2(\frac{{{\mathrm{CPM}}}_{i,j}}{10}+1)\), where CPMi,j refers to counts-per-million for gene i in sample j. We divided the TPM/CPM values by 10 as the size of single-cell libraries is estimated to be in the order of 100,000 transcripts, and we, therefore, would like to avoid inflating the expression levels by counting each transcript ~10 times. Following these normalization steps, initial filtering of low-quality cells was done based on low number of detected genes (less than 200 genes) or high expression of mitochondrially encoded genes (more than 3%). The majority of low-quality cells were removed downstream in case they could not be robustly classified as malignant or nonmalignant (see in later section on CNA analysis ‘Classification of cells into malignant or nonmalignant cell types’). Finally, we computed the average expression of each gene i as \(\scriptstyle\log_2((\frac{1}{n}{\sum }_{j=1}^{n}{{\mathrm{TPM|CPM}}}_{i,j}+1))\) and filtered out the lowly expressed genes by limiting the analyzed genes to the top 3,000 most highly expressed genes (using the Seurat v4.0.4 (ref. 43) function CreateSeuratObject). For the cells and genes that passed these quality control filters, we defined relative expression levels by centering the expression levels for each gene across all cells in the dataset as follows: \({{\mathrm{Er}}}_{i,j}={E}_{i,j}-\,\frac{1}{n}{\sum }_{k=1}^{n}{E}_{i,k}\), where n is the number of cells in the dataset.
Clustering and cell-type annotation
To facilitate clustering, cell-type annotation and scoring of droplet-based data, we used the R package Seurat (v4.0.4)43. Unique molecular identifier counts data were normalized and scaled (using the NormalizeData and ScaleData functions) and then clustered (by projecting the data to a lower dimensional space using principal component analysis and then running FindNeighbors and FindClusters functions). Initial classification of tumor-associated macrophages, T cells, oligodendrocytes and endothelial cells was based on the expression of known marker genes16,17,18,19,44, whereas the rest of the cells were annotated as presumed malignant. Following classification of cells as malignant or nonmalignant (see sections ‘Inferring CNAs from gene expression data’ and ‘Classification of cells into malignant or nonmalignant cell types’), the cells classified as nonmalignant were scored (using the AddModuleScore function) for known cell-type signatures and assigned with a cell type using the method explained in the section ‘Assignment of cells to states’. Finally, heterotypic doublets were filtered out using the R package scDblFinder45.
Inferring CNAs from gene expression data
CNAs were estimated as previously described46,47,48,49 using the function infercna from the R package infercna (an implementation of the original method presented in ref. 50, which is available at https://github.com/jlaffy/infercna) with default parameters. Briefly, the algorithm sorts the analyzed genes in each sample according to their chromosomal ___location and applies a moving average with a sliding window of 100 genes within each chromosome to the relative expression values. The scores computed for the cells classified as nonmalignant (oligodendrocytes, macrophages and endothelial cells) define the baseline of normal karyotype, and their average CNA values are used to center the values of all cells.
Classification of cells into malignant or nonmalignant cell types
In cells classified using single-nucleus droplet-based sequencing, we generally encountered lower CNA signal and continuous signal distributions related to (1) lower data quality relative to that observed in single-cell data and (2) inclusion of nonmalignant cell types such as ACs and neurons in the presumed malignant group, as these cell types could not be classified a priori as nonmalignant due to transcriptional similarity with the malignant cells.
We, therefore, used a multistep classification scheme for the droplet-based single-nucleus sequencing data.
Step 1: cell classification by detection of copy-number events
For each cell j, we computed the CNA signal on each chromosome separately as \({{\mathrm{CNA}}}_{j}^{C}=\frac{1}{n}{\sum }_{i=1}^{n}{{\mathrm{CNA}}}_{i,j}^{C}\), where n is the number of genes on chromosome C included in the analysis. We fitted a normal distribution (that is, a classifier) to the signal distributions of the reference cells for each chromosome (using the function fitdistr from the R package MASS v.7.3-56). This enabled assigning each cell with a P value for each chromosome using a two-sided z test (function pnorm from the R package stats v.4.1.1), reflecting whether the cell has a CNA signal that is substantially different than expected from nonmalignant cells. These P values were further corrected for multiple testing (for each chromosome) using the Benjamini–Hochberg (BH) method, and cells with BH-adjusted P value < 0.05 for a particular chromosome were considered as having a copy-number event on that chromosome (either amplification or deletion based on the sign of the CNA signal). Cells with an unadjusted P value < 0.05 for a particular chromosome that failed to achieve statistical significance following adjustment were considered to have a ‘suspicious event’ on that chromosome. Following this, inferred copy-number events were cross-referenced with copy-number calls from WES/WGS data, and only events detected in both methods were considered for classification and were termed ‘real’ events. For a subset of samples, this procedure was not possible due to (1) lack of WES/WGS data or (2) inability to call CNA events from WES/WGS due to low purity/quality. For this subset of cases, we defined a panel of highly recurring CNA events (intersection of highly recurring events inferred from snRNA-seq and WES/WGS data) that were used as a short list of potential events. Cells classified a priori as nonmalignant were reclassified as ‘unresolved’ in case a ‘real’ or ‘suspicious’ CNA event was detected and ‘nonmalignant’ otherwise. Presumed malignant cells with at least one ‘real’ CNA event were classified as ‘malignant’, those with a ‘suspicious’ event were classified as ‘unresolved’ and those with no events at all (‘real’ or ‘suspicious’) were classified as ‘nonmalignant’.
Step 2: exclude questionable cells based on CNA correlation
We define the CNA correlation as the Pearson correlation between each cell and the average CNA profile of the tumor sample the cell originated from. We limit the computation to the chromosomes on which ‘real’ events were detected and a control panel of ~1,000 genes from other chromosomes. Based on the distribution of CNA correlations across the cells classified as Malignant, we defined 0.5 as the lower threshold of malignancy. To determine the upper threshold for nonmalignancy, we performed SNV calling from the snRNA-seq data to estimate the algorithm misclassification rate. Briefly, for each sample with available WES data, we called SNVs for each cell using the Vartrix tool (https://github.com/10XGenomics/vartrix) from the FASTQ files. Misclassified cells were defined as cells harboring a malignant mutation that were classified as nonmalignant in step 1 of the algorithm. Following a cost-effectiveness analysis (misclassification rate versus percentage of excluded cells), we set 0.35 as the upper threshold for nonmalignancy. Finally, cells classified as ‘malignant’ with CNAcor < 0.5 or cells classified as ‘nonmalignant’ with CNAcor > 0.35 were reclassified as ‘Unresolved’ and excluded from downstream analysis.
Step 3: refine classification by setting confidence levels
To refine the classification, we defined confidence levels. Confidence level 1 includes cells classified as malignant that harbor a malignant SNV, cells in which at least 50% of the expected CNA events were detected, including chromosome 7 amplification or chromosome 10 deletion, and cells classified as nonmalignant with CNAcor < 0.25. Confidence level 2 includes cells classified as malignant in which at least 50% of the expected CNA events were detected that do not include chromosome 7 amplification or chromosome 10 deletion, cells classified as malignant in which less than 50% of the expected CNA events were detected, including chromosome 7 amplification or chromosome 10 deletion, and cells classified as nonmalignant with CNAcor < 0.5. The rest of the cells classified as malignant or nonmalignant were assigned confidence level 3.
In the analysis presented in this work, we used only cells in confidence levels 1 and 2.
Deriving metaprograms from gene expression data
To capture the heterogeneity between cells from the same cell type, we leveraged NMF51. NMF was performed on the relative expression values of each sample independently after setting negative values to zero. The NMF algorithm requires defining a priori the ‘k’ parameter, reflecting the expected number of latent features in the data. Because ‘k’ varies between samples and is largely unknown, we ran the NMF algorithm on each sample using different values (3–10), thereby generating 52 programs for each sample. Each of these NMF programs was summarized by the top 50 genes based on the NMF coefficients. Derivation of the metaprograms from the NMF programs is described thoroughly in ref. 24 and will be described here briefly. The method first filters out NMF programs that are not robust (do not recur within the sample it originated from or across several samples) or are redundant within the sample (that is, it highly overlaps with other NMF programs within that sample). Following this, the robust NMF programs are clustered according to the Jaccard similarity using a customized clustering algorithm that iteratively considers the degree of overlap between robust NMF programs and combines the highly overlapping ones to form a cluster. The top 50 most recurring genes in each cluster define a metaprogram.
Overall, the algorithm yielded 16 metaprograms that were annotated based on functional enrichment analysis (using Gene Ontology (GO) terms, mSigDB Hallmark gene sets and gene sets derived from normal brain development datasets). MP 16 included genes from various cell types, resulting in a metaprogram that fits almost all cells and thereby did not reflect any heterogeneity and was therefore excluded from analysis. This same procedure was independently repeated for each nonmalignant cell type.
Assignment of cells to states
Malignant cells were scored for the NMF metaprograms using the Seurat function AddModuleScore. To facilitate cell classification, we generated 20 shuffled expression matrices by sampling 5,000 cells each time and shuffling the expression values for each gene. We then scored each shuffled matrix for the NMF metaprograms, thereby yielding 100,000 normally distributed scores for each expression program. These served as null distributions for cell state classification. For each original cell, we computed a P value for each of the expression programs with a z test (R’s pnorm function) using the statistics of the null distributions that we previously generated. We adjusted all P values for multiple testing using the BH method. Each cell was classified into a specific state if the adjusted P value computed for that state was smaller than 0.05. Cells that achieved an adjusted P < 0.05 for multiple states were assigned to the state for which they scored maximally. Cells that did not achieve an adjusted P < 0.05 for any of the states were assigned an ‘unresolved’ state.
Cells were assigned a ‘cycling’ state on top of their cellular state if they achieved an adjusted P < 0.05 for the cell cycle metaprogram and ‘noncycling’ otherwise.
Hybrids detection and classification
We define hybrids as cells scoring significantly (that is, with adjusted P < 0.05) for at least two cellular states with a difference between the top two significant scores smaller than expected by chance. To quantify the score difference expected by chance, we leveraged the null distribution generated for the state classification algorithm and computed for each artificial cell the difference between the top two states. The 95 quantiles of this score difference distribution (equal to a difference of 0.24) were set as the threshold for determining if a state is singular or hybrid.
To estimate the expected frequency of technical hybrids, we first computed for each pair of states, \(\left(A,{B}\right)\), the expected frequency of hybrids, defined as \({\mathrm{Exp}}\left(A,B\right)={\mathrm{Freq}}\left(A\right)\times {\mathrm{Freq}}\left(B\right)\), and the O/E ratio defined as \({\log }_{2}(\frac{{\mathrm{Obs}}(A,{B})}{{\mathrm{Exp}}(A,B)})\). As this is an overestimation of the expected frequency attributed to technical effects, we estimated this technical factor by averaging across O/E ratios of hybrid pairs detected less than expected by chance (that is, O/E < 0). Following this, we computed the expected frequency of technical hybrids for each hybrid pair by multiplying the expected frequency with the technical factor and defined the O/tE ratio as \({\log }_{2}(\frac{{\mathrm{Obs}}(A,{B})}{{\mathrm{TF}}\times {\mathrm{Exp}}(A,B)})\). Finally, we filtered out insignificant hybrid pairs with large O/tE ratio (attributed to small numbers) using a one-sided Fisher test. The hybrid lineage model was generated using the R package igraph with cell states as nodes and edges connecting the states that form significant hybrid pairs. We did not include the ‘cycling’ state in this analysis because we don’t view ‘cycling’ as an independent state but rather as an additional feature that cells can have on top of their neurodevelopmental identity (that is, AC-like, OPC-like, etc.).
Baseline profile derivation
Each tumor sample was decomposed into seven pseudobulk profiles (one for each of the common states—AC-like, MES-like, hypoxia, GPC-like, OPC-like, NPC-like and NEU-like—given at least 25 cells classified to the particular state) by averaging across the normalized unique molecular identifier counts and log2-transforming. Genes were included if their mean log2-expression value across all pseudobulks was at least 1 and if their median variance (computed separately within state) was at least 2.5. Overall, 1,005 genes passed these filters. Following this, the pseudobulks were analyzed within the state to remove the state-specific signal using principal component analysis. We then derived six gene programs from each state (top and bottom 50 genes of the first three principal components), computed the Jaccard similarity index between each pair of programs and clustered the similarity matrix using hierarchical clustering. This revealed five distinct clusters from which we derived five consensus signatures by including genes that recurred in at least 25% of programs in each of the clusters (with a hard minimum of at least three times). By manual inspection and GO enrichment, we annotated the consensus signatures. Two signatures were excluded due to short length or high suspicion of reflecting a technical artifact. To exclude the possibility that ambient RNA has a role in driving the baseline profiles, we estimated per sample using the R package SoupX the contamination by ambient RNA. There was no difference in the estimated contamination level across the different baseline profiles as well as no significant overlap between the topmost contaminated genes per sample and the metaprogram or baseline profile signatures (data not shown due to lack of significance).
Assigning tumors to composition groups
To measure the differences in tumor composition, we generated for each tumor sample a composition vector reflecting the proportion of each cell type in the tumor sample. This enabled assigning tumors to the following three main composition groups: HMF (percentage of malignant cells >75%), IMF (percentage of malignant cells, 50–75%) and LMF (malignant cells <50%), where the LMF group could be further subdivided according to the dominant TME cell types (percentage of TAM/oligodendrocyte/glio-neural >40%, or mixed in case of no dominant TME cell type). Similarly, we defined proportion vectors for the malignant cell states and assigned tumors to six groups according to the dominant cell state (at least 25% of cells assigned to the particular state)—AC, MES/hypoxia, GPC, OPC/NPC, neuron and mixed (in case of no dominant state). Tumors were classified according to the baseline profile with a maximal score and to the mixed category if the maximal score was less than 0.25.
Multilayer group definition
The association between each pair of features (A, B) was computed using a binomial test with the number of times the pair was observed as the number of successes, the number of tumor samples as the number of experiments and the frequency of feature A times the frequency of feature B as the expected probability of success (under the assumption that the features are independent). We then generated an undirected graph by defining each feature as a node and connecting the nodes with edges in case a statistically significant association (unadjusted for multiple testing) between the two features was observed.
Measuring transcriptional distance between matched pairs
To measure the transcriptional distance between matched pairs, we generated state-controlled pseudobulk profiles from each pair. State control was achieved by downsampling both samples to 25 cells from each state, given that both samples contain at least 25 cells from the particular state, resulting in samples that contain the same number of cells and are balanced across the different gene expression profiles of the cellular states. Following this, the pseudobulk profiles were computed by averaging for each gene across the selected cells, resulting in two pseudobulk profiles. Finally, the Euclidean distance between the two vectors was computed and served as the metric for transcriptional distance.
Survival analysis
Survival analysis was performed using the R packages survival v.3.3-1 and survminer v.0.4.9. Kaplan–Meier estimates were computed using the survfit function and plotted using the ggsurvplot function (survminer package). Statistical significance of the difference between the survival curves was computed using the log-rank test (implemented in the function survdiff). Cox regression analysis and forest plots were performed and plotted using the same packages (coxph and ggforest functions).
Differentially expressed genes between MGMT-low and MGMT-high pairs
To compute the differentially expressed genes between MGMT-low and MGMT-high pairs, we first computed the relative gene expression across time points in each group (that is, longitudinal expression difference), which is defined for each gene i as the average expression of the gene across all T2 samples in the group minus the average gene expression across all T1 samples in the group. Following, we computed the Euclidean distance between these two vectors. MGMT-low genes were defined as the top 100 genes with the largest distance between the groups and positive longitudinal expression difference in the MGMT-low group and negative longitudinal expression difference in the MGMT-high group. MGMT-high genes were defined as the top 100 genes with the largest distance between the groups and positive longitudinal expression difference in the MGMT-high group and negative longitudinal expression difference in the MGMT-low group.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Processed snRNA-seq data generated for this study are available at the Gene Expression Omnibus under accession GSE274546 (10x Genomics). All de-identified somatic mutation calls, copy-number alteration calls and genomic analysis tables are available via Synapse (https://www.synapse.org/care_glioblastoma). Raw sequencing data are available with limitations in accordance with consent forms from the Data Use Oversight System (DUOS) at https://duos.broadinstitute.org/ under the following IDs: DUOS-000475, DUOS-000476, DUOS-000477, DUOS-000478, DUOS-000479 and DUOS-000480.
Code availability
Analysis scripts used in this study are available at GitHub (https://github.com/dravishays/GBM-CARE-WT) and Zenodo52 (https://doi.org/10.5281/zenodo.14966015). Scripts for processing DNA-sequencing data can be accessed at https://github.com/Kcjohnson/care-glass.
References
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Hegi, M. E. et al. Gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 352, 997–1003 (2005).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
Mu, Q. et al. Identifying predictors of glioma evolution from longitudinal sequencing. Sci. Transl. Med. 15, eadh4181 (2023).
Kim, J., Lee, I., Cho, H. J., Lee, J. & Park, P. J. Spatiotemporal evolution of the primary glioblastoma genome article spatiotemporal evolution of the primary glioblastoma genome. Cancer Cell 28, 318–328 (2015).
Kim, H. et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 25, 316–327 (2015).
Barthel, F. P. et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 576, 112–120 (2019).
Kim, K. et al. Integrated proteogenomic characterization of glioblastoma evolution. Cancer Cell 42, 358–377 (2024).
Varn, F. S. et al. Glioma progression is shaped by genetic evolution and microenvironment interactions. Cell 185, 2184–2199.e16 (2022).
Hoogstrate, Y. et al. Transcriptome analysis reveals tumor microenvironment changes in glioblastoma. Cancer Cell 41, 678–692 (2023).
Wang, J. et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 48, 768–776 (2016).
Garofano, L. et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2, 141–156 (2021).
Wang, L. et al. The phenotypes of proliferating glioblastoma cells reside on a single axis of variation. Cancer Discov. 9, 1708–1719 (2019).
Wang, L. et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat. Cancer 3, 1534–1552 (2022).
Nomura, M. et al. The multilayered transcriptional architecture of glioblastoma ecosystems. Nat. Genet. https://doi.org/10.1038/s41588-025-02167-5 (2025).
Campbell, J. N. et al. A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci. 20, 484–496 (2017).
Habib, N. et al. Div-Seq: single-nucleus RNA-seq reveals dynamics of rare adult newborn neurons. Science 353, 925–928 (2016).
Polioudakis, D. et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron 103, 785–801.e8 (2019).
Nowakowski, T. J. et al. Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323 (2017).
Johnson, B. E. et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014).
Kocakavuk, E. et al. Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. Nat. Genet. 53, 1088–1096 (2021).
Kinker, G. S. et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat. Genet. 52, 1208–1218 (2020).
Greenwald, A. C. et al. Integrative spatial analysis reveals a multi-layered organization of glioblastoma. Cell 187, 2485–2501.e26 (2024).
Gavish, A. et al. Hallmarks of transcriptional intratumour heterogeneity across a thousand tumours. Nature 618, 598–606 (2023).
Jin, S. et al. Inference and analysis of cell–cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Gjørlund, M. D. et al. Neuroligin-1 induces neurite outgrowth through interaction with neurexin-1β and activation of fibroblast growth factor receptor-1. FASEB J. 26, 4174–4186 (2012).
Venkatesh, H. S. et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537 (2017).
Müller, T. et al. Neuregulin 3 promotes excitatory synapse formation on hippocampal interneurons. EMBO J. 37, e98858 (2018).
Venkatesh, S. et al. Electrical and synaptic integration of glioma into neural circuits. Nature 573, 539–545 (2019).
Venkataramani, V. et al. Glioblastoma hijacks neuronal mechanisms for brain invasion. Cell 185, 2899–2917.e31 (2022).
Rivera, A. L. et al. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro Oncol. 12, 116–121 (2010).
Butler, M. et al. MGMT status as a clinical biomarker in glioblastoma. Trends Cancer 6, 380–391 (2020).
Costello, J. F., Futsched, B. W., Tanoil, K., Graunkes, D. M. & Pieper, R. Graded methylation in the promoter and body of the 06-methylguanine DNA methyltransferase (MGMT) gene correlates with MGMT expression in human glioma cells. J. Biol. Chem. 269, 17228–17237 (1994).
Touat, M. et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523 (2020).
Alexandrov, L. B., Kim, J., Haradhvala, N. J. & Huang, M. N. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Johnson, K. C. et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat. Genet. 53, 1456–1468 (2021).
Telarovic, I., Wenger, R. H. & Pruschy, M. Interfering with tumor hypoxia for radiotherapy optimization. J. Exp. Clin. Cancer Res. 40, 197 (2021).
Wang, Q. et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56.e6 (2017).
Slyper, M. et al. A single-cell and single-nucleus RNA-seq toolbox for fresh and frozen human tumors. Nat. Med. 26, 792–802 (2020).
Mathur, R. et al. Glioblastoma evolution and heterogeneity from a 3D whole-tumor perspective. Cell 187, 446–463.e16 (2024).
Islam, S. M. et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genom. 2, 100179 (2022).
Shinde, J. et al. Palimpsest: an R package for studying mutational and structural variant signatures along clonal evolution in cancer. Bioinformatics 34, 3380–3381 (2018).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Velmeshev, D. et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019).
Germain, P., Lun, A., Meixide, C. G., Macnair, W. & Robinson, M. D. Doublet identification in single-cell sequencing data using scDblFinder. F1000Res. 10, 979 (2021).
Neftel, C. et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e21 (2019).
Tirosh, I. et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016).
Venteicher, A. S. et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 (2017).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014).
Gaujoux, R. & Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367 (2010).
Spitzer, A., Johnson, K., Nomura, M. & Garofano, L. The multi-layered transcriptional architecture of glioblastoma ecosystems. Zenodo https://doi.org/10.5281/zenodo.14966015 (2025).
Acknowledgements
We thank R.A. DePinho, D. Arons and K. Knight for their support with the GBM CARE initiative. M.L.S. is supported by the GBM CARE grant (supplemented to P30 CA006516-56S6), NIH–NCI grants (R01CA276765, R37CA245523, R01CA258763 and P50CA165962), the Mark Foundation Emerging Leader Award, the MGH Research Scholars Award, the Sontag Foundation Distinguished Scientist Alumni Award and the National Foundation for Cancer Research. I.T. is supported by an ERC consolidator grant (101044318), the Zuckerman STEM Leadership Program and the Mexican Friends New Generation grant and is the incumbent of the Dr. Celia Zwillenberg-Fridman and Dr. Lutz Zwillenberg Career Development Chair. A.I. is supported by NIH grants (R35CA253183, P30CA013696-46S1 and P30CA240139). R.G.W.V. is supported by NIH grants R01CA237208, R21NS114873, R21CA256575 and P30CA034196. A. Lasorella is supported by NIH grants (R01CA268592, R01CA280560 and R01CA239721). M.N. is supported by the Japan Society for the Promotion of Science Overseas Research Fellowship from the Japan Society for the Promotion of Science and an Overseas Research Fellowship from the Uehara Memorial Foundation. A.S. is partially supported by the Israeli Council for Higher Education via the Weizmann Data Science Research Center. A. Lipsa, A.G. and S.P.N. are supported by the Luxembourg National Research Fund and TRANSCAN3 grants (C20/BM/14646004/GLASSLUX and INTER/TRANSCAN22/17612718/PLASTIG). L.N.G.C. has received honoraria from Elsevier, BMJ Best Practice, Oakstone Publishing and Servier, as well as research support from Merck & Co., Conquer Cancer (the American Society of Clinical Oncology Foundation), the Robert Wood Johnson Foundation and the National Cancer Institute. We thank the PROACTIVE team at MD Anderson for their assistance in procuring patient material. We gratefully acknowledge the contribution of the Jackson Laboratory Single Cell Biology service, S. Bessonett, in particular, the Genome Technologies service and Cyberinfrastructure high-performance computing resources at the Jackson Laboratory for expert assistance related to processing NORLUX/SNU samples and data analysis. These shared services are supported in part by the Jackson Laboratory Cancer Center (P30CA034196). We acknowledge the contribution of R. Matsuura in the Neurosurgery Laboratory, Tokyo University Hospital, for the technical assistance in preparing clinical samples. We thank SciStories for the figure schematics. We also thank the patients and their families for their generous donations to biomedical research.
Author information
Authors and Affiliations
Contributions
A.S., K.C.J., M.N., L.G., A. Lasorella, A.I., R.G.W.V., I.T. and M.L.S. conceived and designed the project. M.N., F.S.V., K.J.A. and Y.T.O. led the data generation. A.S., K.C.J., L.G. and I.T. performed computational analyses. D.N., N.G.D., A.C.G., L.B., F.S.V., F.D., S.G., K.J.A., S.M., F.D., L.N.G.C., T.C., N.R., C.R. and J.B.P. provided experimental and analytical support. A. Lipsa, F.H., A.G., S.P.N., L.N., S.K., S.D., H.E.M., S.H.P., F.B., A. Laurenge, A.L.D.S., B.M., A.P., M.S., S.T., N.S., D.M.A., S.T.K., J.T.H., W.K.A.Y. and K.L.L. provided patient samples and curated the clinical annotation. A.S., K.C.J., M.N., L.G., A. Lasorella, A.I., R.G.W.V., I.T. and M.L.S. wrote the paper, with feedback from all authors. A.I., A. Lasorella, R.G.W.V., I.T. and M.L.S. jointly supervised this work and interpreted the results.
Corresponding authors
Ethics declarations
Competing interests
M.L.S. is an equity holder, scientific cofounder and advisory board member of Immunitas Therapeutics. I.T. is an advisory board member of Immunitas Therapeutics and a scientific cofounder of Cellyrix. R.G.W.V. is a cofounder and equity holder in Boundless Bio. The authors declare that such activities have no relationship to the present study. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Richard Gilbertson, Wolfgang Wick and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Longitudinal profiling of a clinically annotated GBM cohort.
a, Number of cells per sample (post QC) across the 3 time points. Each point represents a sample. There are no statistically significant differences between the groups (tested with two-sided Kruskal–Wallis test, p = 0.51). b, Average number of detected genes per sample (post QC) across the 3 time points. Each point represents a sample. There are no statistically significant differences between the groups (tested with two-sided Kruskal–Wallis test, p = 0.27). c, Overview of SNV and CNA in the GBM cohort. Each column represents a single patient with longitudinal DNA data and a matched normal blood sample. The top panel indicates the total proportion of mutations that are either detected across both T1 and T2 (that is, shared), only in T1 or only in T2, which are sorted by decreasing shared mutation fraction. The middle panel represents the mutation variant status for driver genes in GBM, while the bottom panel presents the driver CNAs.
Extended Data Fig. 2 Longitudinal cell-type changes in the TME, global similarity of cell states between primary and recurrent samples and prognostically significant cell-type profiles.
a, Relative granular cell-type abundance across the two time points (primary and recurrence). Unadjusted two-sided paired Wilcoxon rank-sum test is shown (n = 56 patients). Dotted lines connect patient values. b, Relative granular non-malignant cell state abundance across the two time points (primary and recurrence) that showed statistical significance between time points following adjustment for multiple comparisons. Unadjusted two-sided paired Wilcoxon rank-sum test is shown (n = 56 patients). Dotted lines connect patient values. c, Average proportion of cellular composition (left) of malignant cell state composition (middle) and average baseline profile score (right) for each feature group. d, Available clinical data information for this study and ref. 14 dataset. e, Comparison of the number of detected genes per cell in cells passing quality filters between this study and ref. 14 dataset. Two-sided Wilcoxon rank-sum test is shown. Box plots span from the first to third quartiles, median values are indicated by a horizontal line, whiskers show 1.5× interquartile range and outlier points are not shown. Continuous distributions of all values are shown to the right of each box plot. f, Proportion (%) of samples from ref. 14 assigned to each compositional group across the two time points (primary and recurrence). g, Kaplan–Meier curves depicting the survival time after 2nd resection according to the transcriptomic features at 2nd resection, which shows the statistical significance in the distribution between groups. Top panel is for composition group. Bottom panel is for malignant state groups. Statistical significance of survival difference between the groups in each panel was computed using the log-rank test. h, Forest plot reflects Cox proportional hazard model for time from 2nd resection to outcome, including relevant biological and clinical covariates in addition to composition group (top) and malignant state group (bottom). Each variable is presented along with its categorical possibilities and sample size on the left, while the boxes represent the hazard ratio and lines represent the 95 % confidence interval.
Extended Data Fig. 3 Malignant state abundance and malignant-TME interactions across time points.
a,b, Malignant cell state abundance across time points. Dotted lines connect patient values. Unadjusted two-sided paired Wilcoxon rank-sum tests are shown. a, This study (n = 50 patients with at least 50 malignant cells in both time points). b, ref. 14 dataset. (n = 25 patients). c, Proportion (%) of tumors in each malignant group in the ref. 14 dataset. d, Box plots of the average BP scores per sample across the two time points. e, Interaction graph of associations between features conserved across time points. Edges represent statistically significant (p < 0.05) Pearson correlation in both time points. f, Heatmap of ligand–receptor cross-talks. Each row represents a ligand–receptor interaction, while each column represents a non-malignant cell type—malignant cell state pair, such that the ligand is expressed by the corresponding non-malignant cell type and the receptor by the malignant cell state. The colors represent the log(odds ratio) from the two-sided Fisher’s exact test for checking whether the ligand–receptor interaction is significantly present in T2 (log(odds ratio >0), purple scale) or T1 (log(odds ratio <0), orange scale). Only significant interactions are reported in the heatmap (**p < 0.01, *p < 0.05).
Extended Data Fig. 4 Divergence of individual sample pairs.
a, Sankey plots depicting the transitions between time points across malignant states for ref. 14 dataset. b, Representative two pairs that showed opposite longitudinal transcriptomic changes in the two pairs profiled at two time points highlight unpredictable gene expression trajectories. Heatmap representing three transcriptomic layers between time points are shown.
Extended Data Fig. 5 MGMT expression in GBM single cells influences recurrence trajectories.
a, Surgical interval for the patients included in this analysis stratified by the MGMT promoter methylation status. X axis reflects the time from 1st to 2nd resection. Statistical significance of survival difference between the groups in each panel was computed using the log-rank test. b, Surgical interval analysis for the IDH-wildtype patients in the GLASS cohort stratified by the MGMT promoter methylation status. X axis reflects the time from 1st to 2nd resection. Statistical significance of survival difference between the groups in each panel was computed using the log-rank test. c, Logistic regression model using the average of MGMT gene expression in primary samples to infer the MGMT methylation status in the ref. 14 dataset. d, Proportion (%) of each cell type across the two time points (primary and recurrence) stratified by MGMT expression level. For d–g, box plots represent the median value as a solid horizontal line, lower and upper edges correspond to the first and third quartiles (the 25th and 75th percentiles), and the whiskers represent plus and minus 1.5× the interquartile range. Individual sample values are shown as jittered points. e, Proportion (%) of each malignant cell state across the two time points (primary and recurrence) stratified by MGMT expression level. f, Difference within patients in proportion (% in R − % in P) of each cell type across MGMT expression levels. g, Difference within patients in BP scores (score in R − score in P) across MGMT expression levels. h, Differentially expressed genes between MGMT-low and high groups across the recurrence trajectories (Methods). Each dot represents a gene, and dots are colored according to the group of DE genes to which they belong. i, CONS-UP and CONS-DN signatures plotted for patients included in the analysis. Value represents the T2–T1 difference (log2(FC) of gene expression) for each patient. Patients are ordered according to the difference between signature scores. Genes are ordered according to their correlation (across patients) with the difference between signature scores.
Extended Data Fig. 6 Mutational signatures associated with recurrence trajectories.
a, Mutation burden at both time points for patients with matched normal blood DNA (n = 46 samples per time point, two-sided paired Wilcoxon test). b, COSMIC v3 mutational signature contribution for two patients with high mutation burden at both time points with minimal contribution from SBS11 (alkylating agent signature). c, Selected single base substitution (SBS) mutational signature proportions for GLASS IDH-wildtype cases for both time point 1 and time point 2. Dotted lines connect samples from the same patients. SBS1 (aging/clock-like etiology), SBS11 (alkylating agent) and SBS21 (defective mismatch repair). P values represent two-sided paired Wilcoxon rank-sum test. d, Longitudinal difference in small insertion burden between T1 and T2 with dotted lines connecting patient values. Analysis restricted to non-hypermutant cases. P-value reflects two-sided paired Wilcoxon rank-sum test. e, Longitudinal change (T2–T1) in the hypoxia cell state abundance is plotted on the y axis against the small deletion burden specific to the recurrent tumor with the linear regression line shown in blue. The Pearson correlation coefficient and P value are presented. f, Longitudinal change in the hypoxia and MES-like cell state relative abundances for patients that acquired SBS21 mutations but that did not acquire small deletion burden increase. g, Bulk RNA data from the GLASS consortium were scored for the MES-like snRNA malignant metaprogram signature, and the relative single-sample gene set enrichment analysis (ssGSEA) score is presented on the y axis. The panel shows the longitudinal change in the MES-like signature for those patients whose tumor had a small deletion increase. P value from paired Wilcoxon rank-sum test. h,i, Scatterplots that represent the scaled genetic distance for SNVs (proportions of private mutations) h, and CNAs (differences in copy-number altered regions, i, versus scaled transcriptional distance (snRNA). Shown is the Pearson’s correlation coefficient, associated P value, with the linear regression line shown in blue for both h and i.
Supplementary information
Supplementary Table 1
Cohort description, including sample source, sequencing platforms, patients’ genetic and clinical metadata. Lab, laboratory; M, male; F, female; NA, not accessed; CCNU, lomustine; ACNU, nimustine hydrochloride.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Spitzer, A., Johnson, K.C., Nomura, M. et al. Deciphering the longitudinal trajectories of glioblastoma ecosystems by integrative single-cell genomics. Nat Genet 57, 1168–1178 (2025). https://doi.org/10.1038/s41588-025-02168-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-025-02168-4
This article is cited by
-
The multilayered transcriptional architecture of glioblastoma ecosystems
Nature Genetics (2025)
