
Abstract
A paradigm shift in drug development is the discovery of small molecules that harness the ubiquitin-proteasomal pathway to eliminate pathogenic proteins. Here we provide a modality for targeted protein degradation in lysosomes. We exploit an endogenous lysosomal pathway whereby protein arginine methyltransferases (PRMTs) initiate substrate degradation via arginine methylation. We developed a heterobifunctional small molecule, methylarginine targeting chimera (MrTAC), that recruits PRMT1 to a target protein for induced degradation in lysosomes. MrTAC compounds degraded substrates across cell lines, timescales and doses. MrTAC degradation required target protein methylation for subsequent lysosomal delivery via microautophagy. A library of MrTAC molecules exemplified the generality of MrTAC to degrade known targets and neo-substrates—glycogen synthase kinase 3β, MYC, bromodomain-containing protein 4 and histone deacetylase 6. MrTAC selectively degraded target proteins and drove biological loss-of-function phenotypes in survival, transcription and proliferation. Collectively, MrTAC demonstrates the utility of endogenous lysosomal proteolysis in the generation of a new class of small molecule degraders.

This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
269,00 € per year
only 22,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
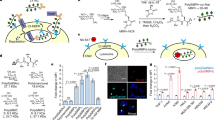

Data availability
All data reporting the findings of this study are included in the Article and within the source data found in the Supplementary Information. Proteomics data have been submitted to the ProteomeXchange Consortium in the PRIDE partner repository with the dataset identifier PXD054653. Source data are provided with this paper.
References
Balch, W. E., Morimoto, R. I., Dillin, A. & Kelly, J. W. Adapting proteostasis for disease intervention. Science 319, 916–919 (2008).
Li, K. & Crews, C. M. PROTACs: past, present and future. Chem. Soc. Rev. 51, 5214–5236 (2022).
Spradlin, J. N., Zhang, E. & Nomura, D. K. Reimagining druggability using chemoproteomic platforms. Acc. Chem. Res. 54, 1801–1813 (2021).
Békés, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 (2022).
Liu, X. & Ciulli, A. Proximity-based modalities for biology and medicine. ACS Cent. Sci. 9, 1269–1284 (2023).
Sakamoto, K. M. et al. Protacs: chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001).
Chirnomas, D., Hornberger, K. R. & Crews, C. M. Protein degraders enter the clinic—a new approach to cancer therapy. Nat. Rev. Clin. Oncol. 20, 265–278 (2023).
Schneider, M. et al. The PROTACtable genome. Nat. Rev. Drug Discov. 20, 789–797 (2021).
Weng, G. et al. PROTAC-DB 2.0: an updated database of PROTACs. Nucleic Acids Res. 51, D1367–D1372 (2023).
Bashore, C. et al. Targeted degradation via direct 26S proteasome recruitment. Nat. Chem. Biol. 19, 55–63 (2023).
Ali, E. M. H., Loy, C. A. & Trader, D. J. ByeTAC: bypassing an E3 ligase for targeted protein degradation. Preprint at bioRxiv https://doi.org/10.1101/2024.01.20.576376 (2024).
Takahashi, D. et al. AUTACs: cargo-specific degraders using selective autophagy. Mol. Cell 76, 797–810 (2019).
Ji, C. H. et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat. Commun. 13, 904 (2022).
Fu, Y. et al. Degradation of lipid droplets by chimeric autophagy-tethering compounds. Cell Res. 31, 965–979 (2021).
Li, Z. et al. Allele-selective lowering of mutant HTT protein by HTT–LC3 linker compounds. Nature 575, 203–209 (2019).
Ahn, G. et al. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 17, 937–946 (2021).
Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
Ahn, G. et al. Elucidating the cellular determinants of targeted membrane protein degradation by lysosome-targeting chimeras. Science 382, eadf6249 (2023).
Zhou, Y., Teng, P., Montgomery, N. T., Li, X. & Tang, W. Development of triantennary N-acetylgalactosamine conjugates as degraders for extracellular proteins. ACS Cent. Sci. 7, 499–506 (2021).
Caianiello, D. F. et al. Bifunctional small molecules that mediate the degradation of extracellular proteins. Nat. Chem. Biol. 17, 947–953 (2021).
Wong, E. & Cuervo, A. M. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb. Perspect. Biol. 2, a006734 (2010).
Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl Acad. Sci. USA 116, 358–366 (2019).
Poirson, J. et al. Proteome-scale discovery of protein degradation and stabilization effectors. Nature 628, 878–886 (2024).
Owens, D. D. G. et al. A chemical probe to modulate human GID4 pro/N-degron interactions. Nat. Chem. Biol. 20, 1164–1175 (2024).
Ichikawa, S. et al. The E3 ligase adapter cereblon targets the C-terminal cyclic imide degron. Nature 610, 775–782 (2022).
Makaros, Y. et al. Ubiquitin-independent proteasomal degradation driven by C-degron pathways. Mol. Cell 83, 1921–1935 (2023).
Lucas, X. & Ciulli, A. Recognition of substrate degrons by E3 ubiquitin ligases and modulation by small-molecule mimicry strategies. Curr. Opin. Struct. Biol. 44, 101–110 (2017).
Nabet, B. et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441 (2018).
Albrecht, L. V., Ploper, D., Tejeda-Muñoz, N. & De Robertis, E. M. Arginine methylation is required for canonical Wnt signaling and endolysosomal trafficking. Proc. Natl Acad. Sci. USA 115, E5317–E5325 (2018).
Franco, C. N. et al. Vitamin B6 is governed by the local compartmentalization of metabolic enzymes during growth. Sci. Adv. 9, eadi2232 (2023).
Larsen, S. C. et al. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 9, rs9 (2016).
Bedford, M. T. & Clarke, S. G. Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 (2009).
Dotson, H. L. & Ngo, J. T. SNAP-tag and HaloTag fused proteins for HaSX8-inducible control over synthetic biological functions in engineered mammalian cells. Preprint at bioRxiv https://doi.org/10.1101/2022.08.12.503781 (2022).
Buckley, D. L. et al. HaloPROTACS: use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chem. Biol. 10, 1831–1837 (2015).
Erhart, D. et al. Chemical development of intracellular protein heterodimerizers. Chem. Biol. 20, 549–557 (2013).
Kaidanovich-Beilin, O. & Woodgett, J. R. GSK-3: functional insights from cell biology and animal models. Front. Mol. Neurosci. 4, 40 (2011).
Taelman, V. F. et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148 (2010).
Douglass, E. F., Miller, C. J., Sparer, G., Shapiro, H. & Spiegel, D. A. A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc. 135, 6092–6099 (2013).
Haid, R. T. U. & Reichel, A. A mechanistic pharmacodynamic modeling framework for the assessment and optimization of proteolysis targeting chimeras (PROTACs). Pharmaceutics 15, 195 (2023).
Weibrecht, I. et al. Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev. Proteomics 7, 401–409 (2010).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17, 1–382 (2021).
Sahu, R. et al. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20, 131–139 (2011).
Bishop, N. & Woodman, P. ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol. Biol. Cell 11, 227–239 (2000).
El-Khoueiry, A. B. et al. Phase 1 study of GSK3368715, a type I PRMT inhibitor, in patients with advanced solid tumors. Br. J. Cancer 129, 309–317 (2023).
Kim, E. et al. Protection of c-Fos from autophagic degradation by PRMT1-mediated methylation fosters gastric tumorigenesis. Int. J. Biol. Sci. 19, 3640–3660 (2023).
Hooper, C., Killick, R. & Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 104, 1433–1439 (2008).
Bilic, J. et al. Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316, 1619–1622 (2007).
Clevers, H. & Nusse, R. Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 (2012).
Kress, T. R., Sabò, A. & Amati, B. MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593–607 (2015).
Chen, H., Liu, H. & Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 3, 5 (2018).
Boike, L. et al. Discovery of a functional covalent ligand targeting an intrinsically disordered cysteine within MYC. Cell Chem. Biol. 28, 4–13 (2021).
Belkina, A. C. & Denis, G. V. BET ___domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 12, 465–477 (2012).
Winter, G. E. et al. BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 67, 5–18 (2017).
Young, P., Deveraux, Q., Beal, R. E., Pickart, C. M. & Rechsteiner, M. Characterization of two polyubiquitin binding sites in the 26 S protease subunit 5a. J. Biol. Chem. 273, 5461–5467 (1998).
Hines, J., Lartigue, S., Dong, H., Qian, Y. & Crews, C. M. MDM2-recruiting PROTAC offers superior, synergistic anti-proliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 79, 251–262 (2019).
Zengerle, M., Chan, K.-H. & Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10, 1770–1777 (2015).
Simpson, L. M. et al. Inducible degradation of target proteins through a tractable affinity-directed protein missile system. Cell Chem. Biol. 27, 1164–1180 (2020).
Lee, Y.-S. et al. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 68, 7561–7569 (2008).
Haberland, M., Montgomery, R. L. & Olson, E. N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 (2009).
Bradner, J. E. et al. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238–243 (2010).
Hanswillemenke, A. et al. Profiling the interactome of oligonucleotide drugs by proximity biotinylation. Nat. Chem. Biol. 20, 555–565 (2024).
Ohana, R. F. et al. Deciphering the cellular targets of bioactive compounds using a chloroalkane capture tag. ACS Chem. Biol. 10, 2316–2324 (2015).
Hu, Y. et al. BRD4 inhibitor inhibits colorectal cancer growth and metastasis. Int. J. Mol. Sci. 16, 1928–1948 (2015).
Li, L. et al. Histone deacetylase inhibitor sodium butyrate suppresses DNA double strand break repair induced by etoposide more effectively in MCF-7 cells than in HEK293 cells. BMC Biochem. 16, 2 (2015).
Tran, A. D.-A. et al. HDAC6 deacetylation of tubulin modulates dynamics of cellular adhesions. J. Cell Sci. 120, 1469–1479 (2007).
Perera, R. M. & Zoncu, R. The lysosome as a regulatory hub. Annu. Rev. Cell Dev. Biol. 32, 223–253 (2016).
Miao, Y. et al. Bispecific aptamer chimeras enable targeted protein degradation on cell membranes. Angew. Chem. Int. Ed. 60, 11267–11271 (2021).
Cotton, A. D., Nguyen, D. P., Gramespacher, J. A., Seiple, I. B. & Wells, J. A. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J. Am. Chem. Soc. 143, 593–598 (2021).
Hsia, O. et al. Targeted protein degradation via intramolecular bivalent glues. Nature 627, 204–211 (2024).
Bao, J. et al. Discovery of novel pdeδ autophagic degraders: a case study of autophagy-tethering compound (ATTEC). ACS Med. Chem. Lett. 15, 29–35 (2024).
Wang, L., Klionsky, D. J. & Shen, H.-M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 24, 186–203 (2023).
Nabet, B. et al. Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat. Commun. 11, 4687 (2020).
Wang, W. W. et al. Targeted protein acetylation in cells using heterobifunctional molecules. J. Am. Chem. Soc. 143, 16700–16708 (2021).
Siriwardena, S. U. et al. Phosphorylation-inducing chimeric small molecules. J. Am. Chem. Soc. 142, 14052–14057 (2020).
Hines, J., Gough, J. D., Corson, T. W. & Crews, C. M. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc. Natl Acad. Sci. USA 110, 8942–8947 (2013).
Wu, Q., Schapira, M., Arrowsmith, C. H. & Barsyte-Lovejoy, D. Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 20, 509–530 (2021).
Acknowledgements
We thank members of the Albrecht Lab for their support in this research and thank J. Leonard for assistance in modeling and conceptualization. The research was supported in part by Ono Pharmaceuticals (award to L.V.A.) and we thank the Cystinosis Research Foundation (CRF) for funding. We would like to thank the UCI Mass Spectrometry Facility and B. Katz for assistance with the collection and analysis of protein MS data. Data were collected on a Waters Acquity UPLC Xevo G2-XS QTOF system (NIH supplemental funding support received by J.S. Nowick (National Institute of General Medical Sciences (NIGMS) GM097562), V.Y. Duong (NIH GM105938) and O. Cinquin (NIGMS GM102635)). We would like to thank C. Yu and L. Huang at the UCI High-end Mass Spectrometry Facility for their help and service in MS data acquisition and analysis. We would also like to thank the Comprehensive Liver Research Center at University of California, Los Angeles (UCLA).
Author information
Authors and Affiliations
Contributions
L.V.A., L.J.S. and C.N.F conceived the study. L.V.A. and L.J.S. designed the experiments. L.J.S., C.N.F., M.C., S.T.N., R.L.W., M.F.K. and K.S. contributed to cell biology experiments. C.A.L., J.O., S.R.L. and D.J.T. contributed to compound synthesis. C.F. and J.Z. contributed to CRISPR cell line construction. L.V.A. and L.J.S. analyzed the data. L.V.A. and L.J.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Biology thanks Cheryl Arrowsmith, Yong Tae Kwon and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Expression and degradation of GSK3β by MrTAC.
a, Schematic of types of MrTAC compounds to mediate proximity between a protein target and a protein arginine methyltransferase (PRMT). Indirect MrTAC dimerizes HaloTag and SNAP-tag fusion proteins that are encoded alongside the PRMT and target. b, Representative immunofluorescence of HEK293 cells stably expressing GSK3β-SNAP-mCherry-FLAG (red) and Halo-PRMT1-Myc (green). c, Immunoblot analysis of GSK3β levels (left) and PRMT1 levels (endogenous, Halo-tagged monomer and Halo-tagged in MrTAC complex; right) during either a 24- or 3-h MrTACHaXS8 dose curve in stably expressing HEK293s. d, Confocal microscopy of HEK293T cells either non-transfected or transfected with GSK3β-SNAP-FLAG (red) and Halo-PRMT1-Myc (green; left). Quantification (right) shows proportion of cells expressing both constructs or just one construct relative to the total number of transfected cells over three fields of view. ****P < 0.0001, *P < 0.0217 by one-sided t-test relative to 0% (n = 3 fields of view). e, Immunoblot analysis of endogenous GSK3β and PRMT1, overexpressed GSK3β-SNAP and Halo-PRMT1 and GSK3β-MrTAC-PRMT1 complexes (0.5 µM, 1 h). f, Immunoblot analysis of HEK293T cells transiently expressing Halo-PRMT1 and GSK3β-SNAP in a 24 (left), 3 (middle) or 9-h (right) MrTAC dose curve (top). Bottom graphs show total levels of GSK3β relative to 0 µM. g, Immunoblot analysis of HeLa cells expressing Halo-PRMT1 and GSK3β-SNAP in a 3-h MrTAC dose curve (top). Bottom graph shows total levels of GSK3β relative to 0 µM. Scale bars are 10 µm. Immunoblots are representative of three independent experiments. Data are represented as means ± SEM.
Extended Data Fig. 2 VPS4-mediated GSK3β sequestration during MrTAC treatment.
a, Live-cell confocal microscopy of GSK3β-SNAP-GFP in HeLa cells incubated with Lysotracker over 60 min with 0.5 µM MrTACHaXS8. Image was captured every 10 min. Arrows note colocalization. b, Colocalization of GSK3β-SNAP-FLAG with lysosomal-associated membrane protein (LAMP-1) after MrTACHaXS8 treatment in stable HEK293 cells (0.5 µM, 3 h, mean colocalizing structures over n = 39DMSO and 54MrTAC cells; left) or transiently expressing HEK293T cells (10 µM, 3 h, mean colocalizing structures over n = 24DMSO and 45MrTAC cells; right). Arrows mark colocalization. For either experiment, graphs depict the number of colocalized GSK3β/LAMP-1 puncta within each cell. ****P < 0.0001 and ***P < 0.0010 by unpaired two-sided t-tests. c, Colocalization of HEK293T cells expressing GSK3β-SNAP-FLAG (red) with either wild-type (WT) or dominant negative (DN) VPS4-GFP after MrTACHaXS8 treatment (10 µM, 1 h). d, Colocalization of HeLa cells expressing GSK3β-SNAP-FLAG (red) with either wild-type (WT) or dominant negative (DN) VPS4-GFP after MrTACHaXS8 treatment (0.5 µM, 2 h). e, Immunoblot analysis of target protein levels of siRNA-mediated knockdown of Lamp2a, Atg7 or Vps4a (100 nM) collected on day of experiment completion (n = 3 experiments). Scale bars are 10 µm. Data are represented as means ± SEM.
Extended Data Fig. 3 Methylation of GSK3β by PRMT1.
a, Mass spectrometry of in vitro methylated GSK3β samples in presence (bottom) or absence (top) of PRMT1 and the associated protein coverage by LC–MS QTOF (left). Associated coverage map of GSK3β by LC–MS QTOF (right). b, Mass spectrometry of methylated GSK3β following FLAG immunoprecipitation of MrTAC-treated HEK293 cells that transiently express PRMT1-Halo and GSK3β-SNAP-FLAG (10 µM, 2 h with 400 nM bafilomycin and 10 µM MG132; left). Associated coverage maps of GSK3β and recruited PRMT1 by LC–MS QTOF (right).
Extended Data Fig. 4 MrTAC degradation of SNAP-tagged proteins reshapes cell signaling.
a, Proliferation following MrTACHaXS8 treatment (0.5 µM) in HeLa cells expressing either Halo-PRMT1/GSK3β-SNAP or Halo-empty/SNAP-empty over seven days (n = 4 experiments; left). **P < 0.0051 drug effect and **PD7 < 0.0010 by two-way ANOVA with Bonferroni’s multiple comparisons. Proliferation of non-transfected HeLa cells treated with MrTACHaXS8 at indicated times (n = 8 experiments), ns by unpaired two-sided t-tests (right). b, Immunoblot analysis of PRMT1 protein levels (Halo-tagged and endogenous) in MrTACHaXS8-treated HEK293 cells expressing CRISPR-integrated PRMT1-Halo/GSK3β-SNAP over 3 days (n = 3 experiments). c, Confocal microscopy of c-MYC-SNAP-FLAG colocalization with asymmetric dimethylarginine (ADMA) antibody after MrTAC treatment (1.5 h, 10 µM; left). Thick outlines mark low colocalization, and arrows mark high colocalization. Scale bar is 5 µm. Bars (right) show average Pearson correlation coefficient between FLAG/ADMA channels across all cells marked with DAPI (n = 11 fields of view). ***P < 0.0009 by two-sided unpaired t-test. d, Confocal microscopy of c-MYC-SNAP-FLAG colocalization with lysosomal-associated membrane protein (LAMP-1) in MrTAC-treated cells (1 h, 10 µM; left). Arrows indicate colocalization, scale bar is 10 µm. Bars (right) show average number of colocalized structures within a cell over 9 fields of view as normalized to DMSO condition. **P < 0.0046 by two-sided unpaired t-test. e, Immunoblot analysis of c-MYC-SNAP levels in cells treated with MrTAC (10 µM) at indicated times with cycloheximide (10 µM). Cells were treated in the presence of bafilomycin (100 nM) or MG132 (5 µM; n = 2 experiments). Data are represented as means ± SEM.
Extended Data Fig. 5 Degradation of endogenous targets.
a, Modeling of MrTACJQ1. MrTACJQ1 bound to BRD4 bromodomain 1 (BD1; PDB: 3MXF) and HaloTag (PDB: 6U32). Pink arrow shows direction of PRMT1 fusion (left). BRD4 BD1 binding to MrTACJQ1 compared against JQ1 ligand (gold; middle). HaloTag binding to MrTACJQ1 compared against tetramethylrhodamine-HaloTag (TMR-HT) ligand (gold; right). b, Quantification of immunoblot in Fig 5b by one-way ANOVA with Bonferroni’s multiple comparisons, *P0.1 < 0.0169, **P1 < 0.0076, **P10 < 0.0069. c, Immunoblot analysis of PRMT1-Halo levels through a 4-h MrTACJQ1 treatment (n = 3). d, Quantification of immunoblot in Fig 5f by one-way ANOVA with Bonferroni’s multiple comparisons, *P < 0.0221 (nMrTAC = 4, nControl = 3). e, Immunoblot analysis of PRMT1 levels through a 4-h MrTACSAHA treatment in HEK293s stably expressing PRMT1-Halo (n = 3; left). Right quantifies levels of total PRMT1 (Halo-tagged and monomeric, uncropped blot in source data) relative to 0 µM; ns by one-way ANOVA with Bonferroni’s multiple comparisons. f, Immunofluorescence of PRMT1-Halo in stably expressing HEK293 cells following a 4-h MrTACSAHA treatment with a lysosome-associated membrane protein 1 (LAMP-1) costain; scale bar is 5 µm. g, Analysis of HDAC6 levels through a 4-h MrTACSAHA treatment in HeLa cells transiently expressing PRMT1-Halo by immunoblot (n = 3; left) and immunofluorescence (n = 200 cells; middle); graph shows HDAC6 intensity over total cells marked by DAPI. ****P < 0.0001 by unpaired two-sided t-test (right). h, Immunoblot analysis of HDAC6 levels with MrTACSAHA (1 µM, 4 h) in HEK293s stably expressing PRMT1-Halo co-treated with lysosome inhibitor bafilomycin (400 nM, 2 h pre-treatment) or proteasome inhibitor MG132 (10 µM; n = 3; left). Right, total levels of HDAC6 in +MrTAC cells relative to either group’s -MrTAC condition. *P < 0.0445 by unpaired two-sided t-test. Immunoblots are representative of at least three independent experiments. Data are represented as means ± SEM.
Extended Data Fig. 6 Effect of endogenous MrTAC degradation on cell function.
a, Representative cell density of HEK293 cells stably expressing PRMT1-HaloTag following 6 days of 1 µM MrTACJQ1 or DMSO vehicle (n = 3 experiments); scale bar is 500 µm. b, Immunoblot analysis of Halo-tagged and endogenous PRMT1 levels through a 3-day MrTACSAHA treatment at 1 µM in HEK293s stably expressing PRMT1-Halo (left; n = 3 experiments). Middle and right quantify total levels of PRMT1-Halo and endogenous PRMT1, respectively, as normalized to the DMSO condition, ns by unpaired two-sided t-test. All quantified values are means ± SEM.
Supplementary information
Supplementary Information
Supplementary Note.
Supplementary Video 1
Live-cell confocal microscopy of GSK3β-SNAP-GFP degradation in HeLa cells over 60 min with 0.1 µM MrTAC. Image was captured every 5 min. Scale bar is 5 µm.
Supplementary Video 2
Live-cell confocal microscopy of GSK3β-SNAP-mCherry degradation in stably expressing HEK293 cells incubated with LysoTracker over 40 min with 0.1 µM MrTAC. Image was captured every 6 min. Scale bar is 10 µm.
Supplementary Video 3
Live-cell confocal microscopy of GSK3β-SNAP-GFP degradation in HeLa cells incubated with LysoTracker over 60 min with 0.5 µM MrTAC. Image was captured every 10 min. Scale bar is 10 µm, arrows note colocalization.
Source data
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Unprocessed western blots.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Unprocessed western blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Unprocessed western blots.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 1
Unprocessed western blots.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Unprocessed western blots.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 6
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Seabrook, L.J., Franco, C.N., Loy, C.A. et al. Methylarginine targeting chimeras for lysosomal degradation of intracellular proteins. Nat Chem Biol 20, 1566–1576 (2024). https://doi.org/10.1038/s41589-024-01741-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41589-024-01741-y
This article is cited by
-
Targeted protein degradation for cancer therapy
Nature Reviews Cancer (2025)
