
Abstract
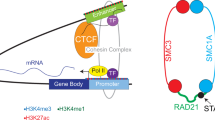
Recent findings indicate that nuclear speckles, a distinct type of nuclear body, interact with certain chromatin regions in a ground state. Here, we report that the chromatin structural factors CTCF and cohesin are required for full ground-state association between DNA and nuclear speckles. We identified a putative speckle-targeting motif (STM) within cohesin subunit RAD21 and demonstrated that the STM is required for chromatin–nuclear speckle association, disruption of which also impaired induction of speckle-associated genes. Depletion of the cohesin-releasing factor WAPL, which stabilizes cohesin on chromatin, resulted in reinforcement of DNA–speckle contacts and enhanced inducibility of speckle-associated genes. Additionally, we observed disruption of chromatin–nuclear speckle association in patient-derived cells with Cornelia de Lange syndrome, a congenital neurodevelopmental disorder involving defective cohesin pathways. In summary, our findings reveal a mechanism for establishing the ground state of chromatin–speckle association and promoting gene inducibility, with relevance to human disease.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
209,00 € per year
only 17,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
Plasmids generated from this study are available from the corresponding author upon request. The genomic datasets generated during this study have been deposited to the Gene Expression Omnibus under accession number GSE248103. The imaging data supporting this study have not been deposited in a public repository because of the file sizes but are available from the corresponding author upon request. Uncropped blots are included in the Supplementary Information.
Code availability
The software, instructions and code generated by this study used for DNA-FISH analysis and analysis of speckle characteristics are available at https://github.com/Chalietia/CellProfiler. The instructions and code used for the analysis of SON TSA-seq data are available at https://github.com/katealexander/TSAseq-Alexander2020/tree/master/genomicBins_DiffBind.
References
Quinodoz, S. A. et al. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757.e24 (2018).
Morimoto, M. & Boerkoel, C. F. The role of nuclear bodies in gene expression and disease. Biology 2, 976–1033 (2013).
Spector, D. L. & Lamond, A. I. Nuclear speckles. Cold Spring Harb. Perspect. Biol. 3, a000646 (2011).
Mor, A. et al. Influenza virus mRNA trafficking through host nuclear speckles. Nat. Microbiol. 1, 16069 (2016).
Kim, J., Venkata, N. C., Gonzalez, G. A. H., Khanna, N. & Belmont, A. S. Gene expression amplification by nuclear speckle association. J. Cell Biol. 219, e201904046 (2019).
Brown, J. M. et al. Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J. Cell Biol. 182, 1083–1097 (2008).
Alexander, K. A. et al. p53 mediates target gene association with nuclear speckles for amplified RNA expression. Mol. Cell 81, 1666–1681.e6 (2021).
Bhat, P. et al. Genome organization around nuclear speckles drives mRNA splicing efficiency. Nature 629, 1165–1173 (2024).
Chen, Y. et al. Mapping 3D genome organization relative to nuclear compartments using TSA-seq as a cytological ruler. J. Cell Biol. 217, 4025–4048 (2018).
Zhang, L. et al. TSA-seq reveals a largely conserved genome organization relative to nuclear speckles with small position changes tightly correlated with gene expression changes. Genome Res. 31, 251–264 (2021).
Wang, Y. et al. SPIN reveals genome-wide landscape of nuclear compartmentalization. Genome Biol. 22, 36 (2021).
Roseman, S. A. et al. DNA methylation insulates genic regions from CTCF loops near nuclear speckles. Preprint at bioRxiv https://doi.org/10.1101/2023.07.26.550645 (2024).
Li, Y. et al. The structural basis for cohesin–CTCF-anchored loops. Nature 578, 472–476 (2020).
Jin, F. et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503, 290–294 (2013).
Gel, B. et al. regioneR: an R/Bioconductor package for the association analysis of genomic regions based on permutation tests. Bioinformatics 32, 289–291 (2016).
Weintraub, A. S. et al. YY1 is a structural regulator of enhancer–promoter loops. Cell 171, 1573–1588.e28 (2017).
Sullivan, K. D., Galbraith, M. D., Andrysik, Z. & Espinosa, J. M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 25, 133–143 (2018).
Karin, M., Liu, Z. & Zandi, E. AP-1 function and regulation. Curr. Opin. Cell Biol. 9, 240–246 (1997).
Ilik, İ. A. et al. SON and SRRM2 are essential for nuclear speckle formation. eLife 9, e60579 (2020).
Alexander, K. A. et al. Nuclear speckles regulate functional programs in cancer. Nat. Cell Biol. https://doi.org/10.1038/s41556-024-01570-0 (2025).
Ahanger, S. H. et al. Distinct nuclear compartment-associated genome architecture in the developing mammalian brain. Nat. Neurosci. 24, 1235–1242 (2021).
Su, J.-H., Zheng, P., Kinrot, S. S., Bintu, B. & Zhuang, X. Genome-scale imaging of the 3D organization and transcriptional activity of chromatin. Cell 182, 1641–1659.e26 (2020).
Beliveau, B. J., Apostolopoulos, N. & Wu, C. Visualizing genomes with oligopaint FISH probes. Curr. Protoc. Mol. Biol. 105, 14.23.1–14.23.20 (2014).
Wutz, G. et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 36, 3573–3599 (2017).
Pugacheva, E. M. et al. CTCF mediates chromatin looping via N-terminal ___domain-dependent cohesin retention. Proc. Natl Acad. Sci. USA 117, 2020–2031 (2020).
Hansen, A. S., Pustova, I., Cattoglio, C., Tjian, R. & Darzacq, X. CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife https://doi.org/10.7554/eLife.25776 (2017).
Huis in ‘t Veld, P. J. et al. Characterization of a DNA exit gate in the human cohesin ring. Science 346, 968–972 (2014).
Haarhuis, J. H. I. et al. A Mediator–cohesin axis controls heterochromatin ___domain formation. Nat. Commun. 13, 754 (2022).
Cheng, H., Zhang, N. & Pati, D. Cohesin subunit RAD21: from biology to disease. Gene 758, 144966 (2020).
Park, D. S. et al. High-throughput Oligopaint screen identifies druggable 3D genome regulators. Nature 620, 209–217 (2023).
Zhou, A., Ou, A. C., Cho, A., Benz, E. J. & Huang, S.-C. Novel splicing factor RBM25 modulates Bcl-x pre-mRNA 5′ splice site selection. Mol. Cell. Biol. 28, 5924–5936 (2008).
Quinodoz, S. A. et al. RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790.e30 (2021).
Lee, D. P. et al. Robust CTCF-based chromatin architecture underpins epigenetic changes in the heart failure stress–gene response. Circulation 139, 1937–1956 (2019).
Horsfield, J. A. Full circle: a brief history of cohesin and the regulation of gene expression. FEBS J. 290, 1670–1687 (2023).
Cuartero, S. et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat. Immunol. 19, 932–941 (2018).
Moore, J. M. et al. Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PLoS ONE 7, e34915 (2012).
Gomez-Velazquez, M. et al. CTCF counter-regulates cardiomyocyte development and maturation programs in the embryonic heart. PLoS Genet. 13, e1006985 (2017).
Birkenbihl, R. P. & Subramani, S. Cloning and characterization of rad21 an essential gene of Schizosaccharomyces pombe involved in DNA double-strand-break repair. Nucleic Acids Res. 20, 6605–6611 (1992).
Rao, S. S. P. et al. Cohesin loss eliminates all loop domains. Cell 171, 305–320.e24 (2017).
Nora, E. P. et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e22 (2017).
Hyle, J. et al. Acute depletion of CTCF directly affects MYC regulation through loss of enhancer–promoter looping. Nucleic Acids Res. 47, 6699–6713 (2019).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Richter, K., Haslbeck, M. & Buchner, J. The heat shock response: life on the verge of death. Mol. Cell 40, 253–266 (2010).
Li, J. et al. An alternative CTCF isoform antagonizes canonical CTCF occupancy and changes chromatin architecture to promote apoptosis. Nat. Commun. 10, 1535 (2019).
Lu, X., Ng, H. & Bubulya, P. A. The role of SON in splicing, development, and disease. Wiley Interdiscip. Rev. RNA 5, 637–646 (2014).
Deardorff, M. A. et al. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 90, 1014–27 (2012).
Borck, G. et al. Incidence and clinical features of X‐linked Cornelia de Lange syndrome due to SMC1L1 mutations. Hum. Mutat. 28, 205–206 (2007).
Gillis, L. A. et al. NIPBL mutational analysis in 120 Individuals with Cornelia de Lange Syndrome and evaluation of genotype–phenotype correlations. Am. J. Hum. Genet. 75, 610–623 (2004).
Kline, A. D. et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat. Rev. Genet. 19, 649–666 (2018).
Kaur, M. et al. Precocious sister chromatid separation (PSCS) in Cornelia de Lange syndrome. Am. J. Med. Genet. A 138A, 27–31 (2005).
Vrouwe, M. G. et al. Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: evidence for impaired recombinational repair. Hum. Mol. Genet. 16, 1478–1487 (2007).
Kervestin, S. & Jacobson, A. NMD: a multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 13, 700–712 (2012).
Haase, M. & Fitze, G. HSP90AB1: helping the good and the bad. Gene 575, 171–186 (2016).
Whitfield, M. L. et al. Stem-loop binding protein, the protein that binds the 3′ end of histone mRNA, is cell cycle regulated by both translational and posttranslational mechanisms. Mol. Cell. Biol. 20, 4188–4198 (2000).
Hu, S., Lv, P., Yan, Z. & Wen, B. Disruption of nuclear speckles reduces chromatin interactions in active compartments. Epigenetics Chromatin 12, 43 (2019).
Merkenschlager, M. & Nora, E. P. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu. Rev. Genom. Hum. Genet. 17, 17–43 (2016).
Braccioli, L. & Wit de, E. CTCF: a Swiss-army knife for genome organization and transcription regulation. Essays Biochem. 63, 157–165 (2019).
Kim, J.-H. et al. De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual-disability syndrome. Am. J. Hum. Genet. 99, 711–719 (2016).
Cuinat, S. et al. Loss-of-function variants in SRRM2 cause a neurodevelopmental disorder. Genet. Med. 24, 1774–1780 (2022).
Kaplanis, J. et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586, 757–762 (2020).
Nguyen, S. C. & Joyce, E. F. Programmable chromosome painting with Oligopaints. Methods Mol. Biol. 2038, 167–180 (2019).
Petrovic, J. et al. Oncogenic notch promotes long-range regulatory interactions within hyperconnected 3D cliques. Mol. Cell 73, 1174–1190.e12 (2019).
Stirling, D. R. et al. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinformatics 22, 433 (2021).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Xu, S., Grullon, S., Ge, K. & Peng, W. Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol. Biol. 1150, 97–111 (2014).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Risso, D., Ngai, J., Speed, T. P. & Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902 (2013).
Acknowledgements
We thank J.-M. Peters for kindly providing us with the CTCF-AID and RAD21-AID HeLa cell lines. S.L.B. acknowledges support from the National Institutes of Health (NIH) grant R35CA263922. B.B.L. acknowledges support from Harvard University startup funds. I.D.K. acknowledges support from NIH grants RO3HD099530, XO1HL145697 and P01HD052860. E.F.J. acknowledges support from NIH grant R35GM128903. Z.G. acknowledges support from the American Heart Association (24PRE1185932) and Penn Developmental Biology Training Grant T32 HD083185. R.J. acknowledges support from NIH grant R35 HL166663 and the Burroughs Wellcome Fund.
Author information
Authors and Affiliations
Contributions
R.Y., K.A.A. and S.L.B. conceived the project. R.Y., S.R., A.P.S., Z.G., K.T., S.C.N. and K.A.A. developed the methodology. R.Y., S.R., Z.G. and A.P.S. performed the investigation. R.Y. visualized the project. R.J., E.F.J., B.B.L., I.D.K. and S.L.B. acquired funding. R.Y. and S.L.B. were responsible for project administration. R.J., E.F.J., B.L., I.D.K. and S.L.B. supervised the research. R.Y., K.A.A. and S.L.B. wrote the original draft of the manuscript; R.Y., K.A.A., S.L.B., E.F.J. and B.B.L. reviewed and edited the final versions.
Corresponding authors
Ethics declarations
Competing interests
B.B.L. has received research funding from Eisai and AstraZeneca and is a shareholder and member of the scientific advisory board of Light Horse Therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Validation of SON Cut&Run.
a-b. Correlation between technical duplicates of designated SON Cut&Run performed in IMR90 (A) or HeLa (B). c-d. Correlation between SON TSA-seq and SON Cut&Run signal over all CDS (C), or SON Cut&Run and MER-FISH (D) in IMR90. e. Correlation between all nuclear speckle proximity data measured by DNA-FISH and SON Cut&Run under same condition obtained in this study.
Extended Data Fig. 2 Quality control of CTCF depletion experiments.
a. Western blot against CTCF in IMR90. (Top) Representative blot, (bottom) quantification. Data represent mean ± SEM from 3 biological replicates; significance by two-sided Student’s t-test. b. RNA-seq normalized counts of designated samples. Data represent mean ± SEM from 3 biological replicates, significance as adjusted p-value calculated by DESeq2. c. Western blot against CTCF in CTCF-AID HeLa cells, at different timepoints after auxin treatment. M represents marker. d. Immunofluorescence against CTCF-eGFP at different timepoints after auxin treatment. e. (Left) Chromosome 11 view of SON Cut&Run signal. (Right) Quantification of the DDB2 gene locus distance to the nearest speckle upon DMSO or auxin treatment in CTCF-AID HeLa cells. Quantification is based on the following DNA-FISH foci counts: DMSO, n = 289; IAA, n = 685. Box plots display the median (center line), 25th–75th percentiles (box), and minima/maxima within 1.5× interquartile range (whiskers), with outliers shown. Statistical significance was determined by two-sided Wilcoxon test. The track shows hg19 genome coordinates, with the y-axis indicating normalized signal relative to IgG. f. Gene locus-speckle distance measured by DNA-FISH in DMSO-treated HeLa cells, ranked from speckle-proximal to distal. (Bottom) DNA-speckle dissociation at specific loci by DNA-FISH or SON Cut&Run. ‘x’: significant; ‘ns’: not significant. SON signal quantified by SON Cut&Run on two biological replicates; DNA-FISH significance as in Fig. 2c. g. Example view and morphological traits of nuclear speckles measured from Immunofluorescence against SON at designated timepoints after auxin treatment, based on the following number of cells for each group: 0 min, n = 459; 30 min, n = 324; 60 min, n = 760; 24 hr, n = 533. h. RNA-seq normalized counts of SON at designated timepoints after auxin treatment.
Extended Data Fig. 3 Quality control of RAD21 depletion experiments.
a. Western blot against RAD21 in RAD21-AID HeLa cells, at different timepoints after auxin treatment. M represents marker. Data represent mean ± SEM from 3 biological replicates; significance by two-sided Student’s t-test. b. Immunofluorescence against RAD21-eGFP (green) at different timepoints after auxin treatment. c. Example view and morphological traits of nuclear speckles measured from Immunofluorescence against speckle marker SON, at designated timepoints after auxin treatment. Quantification based on the following number of cells for each group: 0 min, n = 689; 30 min, n = 733; 60 min, n = 691. d. RNA-seq normalized counts of speckle marker SON, at designated timepoints after auxin mediated depletion of RAD21. Data represent mean ± SEM from 3 biological replicates, significance as adjusted p-value calculated by DESeq2. e. Average SON signal over gene coding sequences (CDS) in DMSO (green) or auxin mediated knockdown (red), ranked from speckle-proximal to distal in HeLa cells.
Extended Data Fig. 4 Disruption of cohesin complex impacts speckle association.
a. Western blot against STAG1 (Left) and STAG2 (right) in HeLa cell line with siRNA against negative control or designated genes. (Top) Representative blot, (bottom) quantification. Data represent mean ± SEM from 3 biological replicates; significance by two-sided Student’s t-test. b. Quantification of UTP18 gene loci distance to closest nuclear speckle upon siRNA knockdown of different cohesin pathway factors. Quantification based on the following number of DNA-FISH foci for each group: siNEG, n = 1310; siSTAG1, n = 756; siSTAG2, n = 739; siWAPL, n = 701. c. Quantification of KCTD20 gene loci distance to closest nuclear speckle upon siRNA knockdown of different cohesin pathway factors. Quantification based on the following number of DNA-FISH foci for each group: siNEG, n = 613; siSTAG1, n = 582; siSTAG2, n = 476; siWAPL, n = 442. d. Western blot against WAPL in HeLa cell line with siRNA against negative control or WAPL. (Top) Representative blot, (bottom) quantification. e. RNA-seq normalized counts of WAPL in HeLa cell line with siRNA against negative control or WAPL. Quantification based on 3 biological replicates for siNEG and 2 for siWAPL. f. Whole chromosome 6,17,19 view of SON Cut&Run data in HeLa cells with siRNA against WAPL or negative control for 48 hr. Track depicts genome coordinates in hg19, and the y-axis depicts normalized signal over IgG. g. Comparison of SPAD sizes in Mb between HeLa cells with or without siRNA knockdown of WAPL. Number of SPAD: Ctrl, n = 1127; siWAPL, n = 800. h. Western blot against RAD21-GFP in HeLa cell line with designated transfectant. (Left) Representative blot, (right) quantification. i. Immunoblots of the immunoprecipitation of endogenous RAD21 from HeLa cells (left) or IMR90 cells (right). Indicated IP samples were run on separate gels and probed separately with RAD21, SMC1, and RBM25 antibodies as indicated.
Extended Data Fig. 5 Disruption of nuclear speckle association hinders the induction of nuclear speckle proximal genes.
a-c. Differential expression analysis of HeLa cells following 24-hour auxin treatment to degrade CTCF (A), 1-hour auxin treatment to degrade RAD21 (B), and 48-hour siRNA-mediated degradation of WAPL (C). In all volcano plots, blue dots indicate significantly down-regulated genes (Log2 fold change < 0, p < 0.01), red dots indicate significantly up-regulated genes (Log2 fold change > 0, p < 0.01), and grey dots indicate non-significant genes. DEG: differentially expressed genes. d. Differential expression analysis of IMR90 cells following 6-hour nutlin-3a treatment, with and without CTCF knockdown. e. Representative Immunofluorescence image and quantification of nuclear speckle traits of IMR90 cells, under designated treatment. Box plots show median (center line), 25th–75th percentiles (box), and minima/maxima within 1.5× interquartile range (whiskers). Data points outside the first and third quartile range are displayed as outliers. Quantification based on the following number of cells for each group: Ctrl, n = 254, Nutlin-3a, n = 318. f. Differential expression analysis of HeLa cells subjected to a 1-hour heat shock treatment, with or without CTCF knockdown. g. Representative Immunofluorescence image and quantification of nuclear speckle traits of HeLa cells, under designated treatment. 37 °C, n = 340, 45 °C, n = 443.
Extended Data Fig. 6 Differentiate speckle-dependent and independent effects of cohesin disruption on gene induction.
a. ΔInducibility analysis comparing auxin-induced CTCF knockdown to DMSO-treated HeLa cells under heat shock conditions. Genes with significant differential conditional effects were grouped by speckle proximity (left) or proximity to loop anchors (right). Box plots show median (center line), 25th–75th percentiles (box), and minima/maxima within 1.5× interquartile range (whiskers). Data points outside the first and third quartile range are displayed as outliers. Based on RNA-seq of 3 biological replicates, with n indicating gene count per group. b. Overlap of heat-responsive genes (Log2FC > 1 under heat shock) with reduced induction level between siRNA mediated CTCF knockdown and SON knockdown HeLa cells. c. Western blot against RAD21-GFP in HeLa cell line with designated plasmid transfection. (Left) Immunoblot, (Right) quantification. Data represent mean ± SEM from 3 biological replicates; significance by two-sided Student’s t-test. d. Immunofluorescence for RAD21-GFP in HeLa cells with designated plasmid transfection. (Left) Representative image; (Right) quantification of transfection efficiency. Data from 5 fields across 3 biological replicates; significance assessed by two-sided Wilcoxon test.
Extended Data Fig. 7 Quality control and transcriptomic profiling donor derived LCLs.
a. Sanger sequencing results of designated derived LCL cells, within RAD21 gene coding region at chr8: 117869560-117869616. b. RNA-seq normalized counts of RAD21 genes in designated healthy individual or patient derived LCLs. Data represent mean ± SEM from 3 biological replicates for two controls and P561 each, significance as adjusted p-value calculated by DESeq2. c. Representative images with DAPI stained nuclei (blue), immunofluorescence of nuclear speckle component SON (green), in LCL cells from healthy donors (Ctrl1, Ctrl2) and patient 561 (P561-1, P561-2). d. Morphological traits of nuclear speckles measured from immunofluorescence against speckle marker SON in designated healthy individual or patient derived LCLs. e. Differential conditional effect under heat shock between two control LCLs from healthy individual. RNA-seq quantifications are based on 3 biological replicates, and box plots show median (center line), 25th–75th percentiles (box), and minima/maxima within 1.5× interquartile range (whiskers). Significance determined by two-sided Wilcoxon test.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yu, R., Roseman, S., Siegenfeld, A.P. et al. CTCF/RAD21 organize the ground state of chromatin–nuclear speckle association. Nat Struct Mol Biol 32, 1069–1080 (2025). https://doi.org/10.1038/s41594-024-01465-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-024-01465-6
