
Abstract
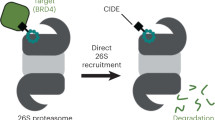
Proteolysis targeting chimeras (PROTACs) hijack E3 ligases and the ubiquitin–proteasome system to achieve selective degradation of neo-substrates. Their ability to target otherwise intractable substrates has rendered them a valuable modality in drug discovery. However, only a handful of over 600 human E3 ligases have been functionalized for PROTAC applications. Here we show that the E3 ligase GID4 (glucose-induced degradation deficient complex 4) can be leveraged for targeted protein degradation using a noncovalent small molecule. We design and synthesize GID4-based PROTACs, exemplified by NEP162, which can eliminate endogenous BRD4 in a GID4- and ubiquitin–proteasome system-dependent manner. NEP162 exhibits antiproliferative activity and inhibits tumor growth in a xenograft model, hinting toward potential anticancer applications. We further present the crystal structures of GID4–PROTAC–BRD4 ternary complexes in three distinct states, unveiling plastic interactions between GID4 and BRD4. These structural insights, combined with in vitro and in vivo data, decipher the molecular basis by which the hereby developed PROTACs recruit BRD4 to GID4 for targeted degradation and expand our arsenal of PROTAC-exploitable E3 ligases.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
27,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
209,00 € per year
only 17,42 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
Atomic coordinates and structure factors have been deposited in the PDB with the accession codes 8X7G (GID4–NEP108–BRD4 ternary complex) and 8X7H (GID4–NEP162–BRD4 ternary complex). The mass spectrometry proteomics data have been deposited at the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository84 with the dataset identifier PXD053449. Source data are provided with this paper.
References
Bekes, M., Langley, D. R. & Crews, C. M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. https://doi.org/10.1038/s41573-021-00371-6 (2022).
Dale, B. et al. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 21, 638–654 (2021).
Scudellari, M. Protein-slaying drugs could be the next blockbuster therapies. Nature 567, 298–300 (2019).
Mullard, A. First targeted protein degrader hits the clinic. Nat. Rev. Drug Discov. https://doi.org/10.1038/d41573-019-00043-6 (2019).
He, M. et al. PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct. Target Ther. 7, 181 (2022).
Chirnomas, D., Hornberger, K. R. & Crews, C. M. Protein degraders enter the clinic—a new approach to cancer therapy. Nat. Rev. Clin. Oncol. 20, 265–278 (2023).
Sakamoto, K. M. et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl Acad. Sci. USA 98, 8554–8559 (2001).
Burslem, G. M. et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem. Biol. 25, 67–77 e63 (2018).
Lai, A. C. & Crews, C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114 (2017).
Henley, M. J. & Koehler, A. N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 20, 669–688 (2021).
Bai, L. et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 36, 498–511 e417 (2019).
Bondeson, D. P. et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015).
Weng, G. et al. PROTAC-DB 2.0: an updated database of PROTACs. Nucleic Acids Res. 51, D1367–D1372 (2023).
Schapira, M., Calabrese, M. F., Bullock, A. N. & Crews, C. M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov. https://doi.org/10.1038/s41573-019-0047-y (2019).
Winter, G. E. et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015).
Lu, J. et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22, 755–763 (2015).
Zengerle, M., Chan, K. H. & Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10, 1770–1777 (2015).
Ishida, T. & Ciulli, A. E3 ligase ligands for PROTACs: how they were found and how to discover new ones. SLAS Discov. https://doi.org/10.1177/2472555220965528 (2020).
Steinebach, C. et al. PROTAC-mediated crosstalk between E3 ligases. Chem. Commun. 55, 1821–1824 (2019).
Girardini, M., Maniaci, C., Hughes, S. J., Testa, A. & Ciulli, A. Cereblon versus VHL: hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 27, 2466–2479 (2019).
Lai, A. C. et al. Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. 55, 807–810 (2016).
Donovan, K. A. et al. Mapping the degradable kinome provides a resource for expedited degrader development. Cell 183, 1714–1731 e1710 (2020).
Bondeson, D. P. et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem. Biol. 25, 78–87 e75 (2018).
Luo, X. et al. Profiling of diverse tumor types establishes the broad utility of VHL-based ProTaCs and triages candidate ubiquitin ligases. iScience 25, 103985 (2022).
Shirasaki, R. et al. Functional genomics identify distinct and overlapping genes mediating resistance to different classes of heterobifunctional degraders of oncoproteins. Cell Rep. 34, 108532 (2021).
Zhang, L., Riley-Gillis, B., Vijay, P. & Shen, Y. Acquired resistance to BET-PROTACs (proteolysis-targeting chimeras) caused by genomic alterations in core components of E3 ligase complexes. Mol. Cancer Ther. 18, 1302–1311 (2019).
Hanzl, A. et al. Functional E3 ligase hotspots and resistance mechanisms to small-molecule degraders. Nat. Chem. Biol. 19, 323–333 (2023).
Lee, J. et al. Discovery of E3 ligase ligands for target protein degradation. Molecules 27, 6515 (2022).
Kobayashi, N. et al. RanBPM, Muskelin, p48EMLP, p44CTLH, and the armadillo-repeat proteins ARMC8alpha and ARMC8beta are components of the CTLH complex. Gene 396, 236–247 (2007).
Francis, O., Han, F. & Adams, J. C. Molecular phylogeny of a RING E3 ubiquitin ligase, conserved in eukaryotic cells and dominated by homologous components, the muskelin/RanBPM/CTLH complex. PLoS ONE 8, e75217 (2013).
Santt, O. et al. The yeast GID complex, a novel ubiquitin ligase (E3) involved in the regulation of carbohydrate metabolism. Mol. Biol. Cell 19, 3323–3333 (2008).
Menssen, R. et al. Exploring the topology of the GID complex, the E3 ubiquitin ligase involved in catabolite-induced degradation of gluconeogenic enzymes. J. Biol. Chem. 287, 25602–25614 (2012).
Liu, H. & Pfirrmann, T. The GID-complex: an emerging player in the ubiquitin ligase league. Biol. Chem. 400, 1429–1441 (2019).
Maitland, M. E. R., Lajoie, G. A., Shaw, G. S. & Schild-Poulter, C. Structural and functional insights into GID/CTLH E3 ligase complexes. Int. J. Mol. Sci. 23, 5863 (2022).
Huffman, N., Palmieri, D. & Coppola, V. The CTLH complex in cancer cell plasticity. J. Oncol. 2019, 4216750 (2019).
Yi, S. A., Sepic, S., Schulman, B. A., Ordureau, A. & An, H. mTORC1-CTLH E3 ligase regulates the degradation of HMG-CoA synthase 1 through the Pro/N-degron pathway. Mol. Cell 84, 2166–2184.e9 (2024).
Gottemukkala, K. V. et al. Non-canonical substrate recognition by the human WDR26-CTLH E3 ligase regulates prodrug metabolism. Mol. Cell 84, 1948–1963 e1911 (2024).
Varshavsky, A. N-degron and C-degron pathways of protein degradation. Proc. Natl Acad. Sci. USA 116, 358–366 (2019).
Chen, S. J., Wu, X., Wadas, B., Oh, J. H. & Varshavsky, A. An N-end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science 355, eaal3655 (2017).
Chen, S. J., Kim, L., Song, H. K. & Varshavsky, A. Aminopeptidases trim Xaa-Pro proteins, initiating their degradation by the Pro/N-degron pathway. Proc. Natl Acad. Sci. USA 118, e2115430118 (2021).
Dong, C. et al. Recognition of nonproline N-terminal residues by the Pro/N-degron pathway. Proc. Natl Acad. Sci. USA 117, 14158–14167 (2020).
Chrustowicz, J. et al. Multifaceted N-degron recognition and ubiquitylation by GID/CTLH E3 ligases. J. Mol. Biol. 434, 167347 (2022).
Dong, C. et al. Molecular basis of GID4-mediated recognition of degrons for the Pro/N-end rule pathway. Nat. Chem. Biol. 14, 466–473 (2018).
Qiao, S. et al. Interconversion between anticipatory and active GID E3 ubiquitin ligase conformations via metabolically driven substrate receptor assembly. Mol. Cell 77, 150–163 e159 (2020).
Kong, K. E. et al. Timer-based proteomic profiling of the ubiquitin-proteasome system reveals a substrate receptor of the GID ubiquitin ligase. Mol. Cell 81, 2460–2476.e11 (2021).
Mohamed, W. I. et al. The human GID complex engages two independent modules for substrate recruitment. EMBO Rep. 22, e52981 (2021).
Sherpa, D. et al. GID E3 ligase supramolecular chelate assembly configures multipronged ubiquitin targeting of an oligomeric metabolic enzyme. Mol. Cell 81, 2445–2459 e2413 (2021).
Chrustowicz, J. et al. Multisite phosphorylation dictates selective E2-E3 pairing as revealed by Ubc8/UBE2H-GID/CTLH assemblies. Mol. Cell 84, 293–308 e214 (2024).
Yazdi, A. K. et al. Chemical tools for the Gid4 subunit of the human E3 ligase C-terminal to LisH (CTLH) degradation complex. RSC Med. Chem. https://doi.org/10.1039/D3MD00633F (2024).
Owens, D. D. G. et al. A chemical probe to modulate human GID4 Pro/N-degron interactions. Nat. Chem. Biol. https://doi.org/10.1038/s41589-024-01618-0 (2024).
Chana, C. K. et al. Discovery and structural characterization of small molecule binders of the human CTLH E3 ligase subunit GID4. J. Med. Chem. 65, 12725–12746 (2022).
Nowak, R. P. et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 14, 706–714 (2018).
Hickey, C. M. et al. Co-opting the E3 ligase KLHDC2 for targeted protein degradation by small molecules. Nat. Struct. Mol. Biol. https://doi.org/10.1038/s41594-023-01146-w (2024).
Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010).
Filippakopoulos, P. et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231 (2012).
Gadd, M. S. et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol. 13, 514–521 (2017).
Schiemer, J. et al. Snapshots and ensembles of BTK and cIAP1 protein degrader ternary complexes. Nat. Chem. Biol. 17, 152–160 (2021).
Mabanglo, M. F. et al. Crystal structures of DCAF1-PROTAC-WDR5 ternary complexes provide insight into DCAF1 substrate specificity. Nat. Commun. 15, 10165 (2024).
O’Boyle, N. M. et al. beta-Lactam estrogen receptor antagonists and a dual-targeting estrogen receptor/tubulin ligand. J. Med. Chem. 57, 9370–9382 (2014).
Farnaby, W. et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 15, 672–680 (2019).
Casement, R., Bond, A., Craigon, C. & Ciulli, A. Mechanistic and structural features of PROTAC ternary complexes. Methods Mol. Biol. 2365, 79–113 (2021).
Chung, C. W. et al. Structural insights into PROTAC-mediated degradation of Bcl-xL. ACS Chem. Biol. 15, 2316–2323 (2020).
Testa, A., Hughes, S. J., Lucas, X., Wright, J. E. & Ciulli, A. Structure-based design of a macrocyclic PROTAC. Angew. Chem. 59, 1727–1734 (2020).
Law, R. P. et al. Discovery and characterisation of highly cooperative FAK-degrading PROTACs. Angew. Chem. 60, 23327–23334 (2021).
Yu, X. et al. A selective WDR5 degrader inhibits acute myeloid leukemia in patient-derived mouse models. Sci. Transl. Med. 13, eabj1578 (2021).
Kofink, C. et al. A selective and orally bioavailable VHL-recruiting PROTAC achieves SMARCA2 degradation in vivo. Nat. Commun. 13, 5969 (2022).
Nayak, D. et al. Development and crystal structures of a potent second-generation dual degrader of BCL-2 and BCL-xL. Nat. Commun. 15, 2743 (2024).
Bachmair, A., Finley, D. & Varshavsky, A. In vivo half-life of a protein is a function of its amino-terminal residue. Science 234, 179–186 (1986).
Koren, I. et al. The eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C-terminal degrons. Cell 173, 1622–1635 e1614 (2018).
Lin, H. C. et al. C-terminal end-directed protein elimination by CRL2 ubiquitin ligases. Mol. Cell 70, 602–613 e603 (2018).
Sherpa, D., Chrustowicz, J. & Schulman, B. A. How the ends signal the end: regulation by E3 ubiquitin ligases recognizing protein termini. Mol. Cell 82, 1424–1438 (2022).
Timms, R. T. et al. A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science 365, eaaw4912 (2019).
Yan, X. et al. Molecular basis for recognition of Gly/N-degrons by CRL2(ZYG11B) and CRL2(ZER1). Mol. Cell https://doi.org/10.1016/j.molcel.2021.06.010 (2021).
Varshavsky, A. N-degron pathways. Proc. Natl Acad. Sci. USA 121, e2408697121 (2024).
Zhang, J. et al. Distinct amino acid-based PROTACs target oncogenic kinases for degradation in non-small cell lung cancer (NSCLC). J. Med. Chem. https://doi.org/10.1021/acs.jmedchem.4c00208 (2024).
Lee, Y. et al. Targeted degradation of transcription coactivator SRC-1 through the N-degron pathway. Angew. Chem. 59, 17548–17555 (2020).
Roth, S. et al. Identification of KLHDC2 as an efficient proximity-induced degrader of K-RAS, STK33, beta-catenin, and FoxP3. Cell Chem. Biol. https://doi.org/10.1016/j.chembiol.2023.07.006 (2023).
Kim, Y. et al. Targeted kinase degradation via the KLHDC2 ubiquitin E3 ligase. Cell Chem. Biol. https://doi.org/10.1016/j.chembiol.2023.07.008 (2023).
Scott, D. C. et al. Principles of paralog-specific targeted protein degradation engaging the C-degron E3 KLHDC2. Nat. Commun. 15, 8829 (2024).
Henning, N. J. et al. Discovery of a covalent FEM1B recruiter for targeted protein degradation applications. J. Am. Chem. Soc. 144, 701–708 (2022).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of COOT. Acta Crystallogr. D 66, 486–501 (2010).
Ma, J. et al. iProX: an integrated proteome resource. Nucleic Acids Res. 47, D1211–D1217 (2019).
Acknowledgements
We thank the staff at beamlines BL19U1 of Shanghai Synchrotron Radiation Facility for assistance in X-ray data collection. This work was supported by National Natural Science Foundation of China grant nos. 82321001 (to C.D.), 32271265 (to C.D.), 82425040 (to L.S.), 82230101 (to L.S.), 22307093 (to D.C.) and 82473144 (to X.Y.), National Youth Top-Notch Talent Support Program in China, Tianjin Municipal Science and Technology Commission grant nos. 22JCZDJC00440 (to C.D.) and 23JCQNJC00540 (to D.C.), Research Foundation of Tianjin Municipal Education Commission grant nos. 2022KJ189 (to D.C.) and 2022KJ191 (to B.Z.), Postdoctoral Fellowship Program and China Postdoctoral Science Foundation (grant nos. GZB20240530 and 2024T170654 to K.B.) and Initiative (grant no. 2024NITFID313) by the National Key Laboratory of Intelligent Tracking and Forecasting for Infectious Diseases. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
C.D., L.S., D.C. and S.X. conceptualized the project and designed experiments. Y.L. performed protein expression, purification and crystallization with help from Q.Z. J.S. and M.Z. performed the synthesis and chemical characterization of all compounds. K.B. carried out the cellular assays. R.G. and J. Zang performed the in vivo assays. X.Y. and J.L. determined the crystal structures. C.D., L.S., D.C., S.X., M.L., J. Zhou, W.M., F.S. and B.Z. analyzed the data. C.D. wrote the paper with critical inputs from all authors.
Corresponding authors
Ethics declarations
Competing interests
C.D., L.S., D.C., J.S. and Y.L. declare the filing of a patent application covering the PROTACs described in this paper. The other authors declare no competing interests.
Peer review
Peer review information
Nature Structural & Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available. Primary Handling Editor: Dimitris Typas, in collaboration with the Nature Structural & Molecular Biology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Design of GID4-based PROTACs for BRD4 degradation.
a, Chemical structures of initially designed GID4-based degraders 1-4. b, RT-qPCR analysis of GID4 mRNA expression level in different cell lines. Data presented are the mean ± S.D. of three independent experiments. c, Immunoblotting analysis of BRD4 degradation in U2OS cells treated with the indicated compounds (2 µM) for 18 h. Representative images, n = 3. d, Pull-down assay using His-GID4 to pull down BRD4BD1 with addition of varying concentrations of NEP108. Representative images, n = 3.
Extended Data Fig. 3 Structural comparison of the GID4-, CRBN- and VHL-recruiting BRD4.
a, Superimposition of GID4-NEP108-BRD4 and CRBN-dBET23-BRD4 (PDB code: 6BN7) ternary complexes against BRD4. For clarity, only one BRD4 model is shown. b, Superimposition of GID4-NEP108-BRD4 and VHL-MZ1-BRD4 (PDB code: 5T35) ternary complexes against BRD4. The ZA loop, BC loop and αC helix involved in the binding are indicated.
Extended Data Fig. 4 Structure-based design of GID4-based degraders.
a, Electrostatic potential surface of NEP108-binding pocket in GID4 and BRD4 (red, negative; blue, positive). b, Chemical structure of the GID4-based NEP179. c, Immunoblotting analysis of BRD4 degradation in U2OS cells treated with the 2 µM NEP179 and NEP162 compounds for 18 h. Representative images, n = 3. d, Immunoblotting analysis of BRD4 proteins in U2OS cells treated with 2 µM NEP162N or NEP162 for 18 h. Representative images, n = 3. e, Concentration-dependent degradation of BRD4 proteins by NEP162 in U2OS cells for 18 h. f, DC50 value of NEP162 for BRD4 in U2OS cells for 18 h. Data are derived from three biologically independent experiments. g,h,i, Immunoblotting analysis of BRD4 degradation in H3122 cells (g), H520 cells (h) and HEC-1-A cells (i) by NEP162 for 18 h. Representative images, n = 3. j, Immunoblotting analysis of BRD4 degradation in SW480 cells treated with 2 µM NEP162 or MZ1 for 18 h. Representative images, n = 3.
Extended Data Fig. 5 Insights into the NEP162-mediated formation of the ternary complex.
a, Pull-down assay using His-GID4 to pull down BRD4BD1 with addition of varying concentrations of NEP162. Representative images, n = 3. b,c, Fo-Fc omit map (green mesh) of the NEP162 (b; J-state and c; L-state) generated before ligand modelling countered at the 1.0 σ level. d, BLI measurements of the interaction of immobilized mutant GID4 (Y168A) and a premix of NEP162-BRD4. Data presented are the mean ± S.D. of three independent experiments. e, BLI measurements of the interaction of immobilized double mutant GID4 (E167A and Y168A) and a premix of NEP162-BRD4. Data presented are the mean ± S.D. of four independent experiments. f, BLI analysis of the binary interaction between GID4 (E167A and Y168A) and NEP162. g, BLI measurements of the interaction of immobilized double mutant GID4 (E167A and Y168A) and a premix of NEP108-BRD4. Data presented are the mean ± S.D. of three independent experiments.
Extended Data Fig. 6 Structural comparison of GID4-based complexes.
a, Overlay of the GID4 structures derived from ternary complexes mediated by NEP108, NEP162 (J-state) and NEP162 (L-state). b, Binding modes of GID4 bound to NEP108, NEP162 (J-state) and NEP162 (L-state). Structures are overlaid in reference to GID4. c, Structural alignment of GID4 in complex with PHRV peptide (PDB code: 6CCU), PSRV peptide (PDB code: 6CD8) and PGLW peptide (PDB code: 6CDC). For clarity, only one GID4 molecule is shown in a surface representation. d, Superimposition of BRD4 structures derived from ternary complexes mediated by NEP108, NEP162 (J-state) and NEP162 (L-state).
Extended Data Fig. 7 Degradation mechanism by NEP162.
a, Analysis of BRD4 protein ubiquitination in U2OS cells pretreated with 10 µM MG132 for 12 h, followed by treatment with DMSO, 2 µM NEP108 or NEP162 for 18 h. b, Immunoblotting analysis of the degradation of natural substrate HMGCS1 in SW480 cells upon Torin1 treatment (200 nM, 16 h) in the presence of cycloheximide (50 ng/L, 16 h). c, Cell viability was conducted in U2OS cells treated with DMSO or increasing concentrations of NEP162 for the indicated times. Data are shown as mean ± S.D. (n = 3 biological replicates); two-sided t-test, *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Extended Data Fig. 8 Comparison of the activity of NEP162 and MZ1 in vivo.
a, Schematic of the administration of tumor-bearing mice. b,c, Images (b) and volumes (c) of tumors isolated from xenograft mice after intraperitoneal injection of DMSO, NEP162N (10 mg/kg), NEP162 (10 mg/kg) or MZ1 (10 mg/kg). Data are shown as mean ± S.D. (n = 5 biological replicates); two-sided t-test, *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. d, Tumor weights of mice at the endpoint. Data are shown as mean ± S.D. (n = 5 biological replicates); two-sided t-test, *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. e, Changes in body weight of mice during the treatments. Data are shown as mean ± S.D. (n = 5 biological replicates). f, Immunofluorescence imaging of TUNEL, and immunohistochemistry imaging of Ki67 and BRD4 in U2OS tumor sections from various groups. g,h,i, Quantitative analysis of tumor sections stained with TUNEL (g), Ki67 (h) and BRD4 (i). Data are shown as mean ± S.D. (n = 5 biological replicates); two-sided t-test, *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Panel a created with BioRender.com.
Supplementary information
Supplementary Information
Supplementary chemistry methods for compounds.
Source data
Source Data Figs. 1–7 and Extended Data Figs. 1, 4, 5, 7 and 8
Statistical source data, all graph data in excel as tabs per figure panel.
Source Data Figs. 1, 3 and 5–7 and Extended Data Figs. 1, 4, 5, 7 and 8
Unprocessed western blots and gels, all uncropped blots and gels as labeled clearly.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Y., Bao, K., Sun, J. et al. Design of PROTACs utilizing the E3 ligase GID4 for targeted protein degradation. Nat Struct Mol Biol (2025). https://doi.org/10.1038/s41594-025-01537-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41594-025-01537-1
