
Abstract
Bladder cancer is one of the leading causes of cancer-related mortality in the urinary system. Understanding genomic information is important in the treatment and prognosis of bladder cancer, but the current method used to identify mutations is time-consuming and labor-intensive. There are now many novel and convenient ways to predict cancerous genomics from pathological slides. However, the publicly available datasets are limited, especially for Asian populations. In this study, we developed a dataset consisting of 75 Asian cases of bladder cancers and 112 Whole-Slide Images with one to two images obtained for each patient. This dataset provides information on the most frequently and clinically significant mutated genes derived by whole-exome sequencing in these patients. This dataset will facilitate exploration and development of novel diagnostic and therapeutic technologies for bladder cancer.
Similar content being viewed by others


Background & Summary
Bladder cancer (BLCA) is one of the most common and fatal malignant tumors of the urinary system1. Bladder cancer genomics aims to understand the genetic basis of tumor cell proliferation and evolution of the cancer genome under mutation and selection by the environment within the body, as well as the immune system and therapeutic interventions2,3,4,5. In medicine, the main reasons for knowing the cumulative phenotypic consequences of somatically acquired genetic, genomic and epigenetic alterations in cancer cells are related to prevention, detection and treatment6,7. For example, the presence of TP53 and ERCC1 mutations might influence the sensitivity of muscle-Invasive bladder cancer (MIBC) to chemotherapy8,9. Furthermore, FGFR3 mutations characterize a subgroup of bladder cancers with a good prognosis, and EGFR, OAS1 and MST1R mutations indicate sensitivity to immunotherapy10,11. The ability to identify these genetic mutations has benefits in terms of treatment of bladder cancer.
Traditionally, acquisition of a cancerous genome requires tumor resection, quality control and sequencing or panel detection, which is time-consuming, labor-intensive and expensive12. Identification of the genomic features of a tumor has significance for treatment and the prognosis13. Numerous studies have attempted to predict the genomic status of tumors from pathological sections or even images14. However, training such models typically requires a large amount of data, and publicly available datasets are scarce15. Most of the current studies are based on The Cancer Imaging Archive (TCIA)16 data and lack publicly available external cohorts for validation, which has a negative impact on the robustness and extensibility of the models. Meanwhile, The Cancer Genome Atlas (TCGA) sequencing data focus predominantly on the Caucasian population, and further validation is needed to determine whether models trained on the TCGA database can be applied to other ethnic groups.
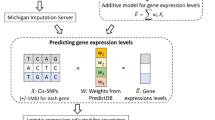
We have previously published a large single-center artificial intelligence study in which the genomic features of 75 Asian patients with muscle-Invasive bladder cancer were successfully predicted by whole-slide images (WSIs)17. Tissues from these patients, all of whom had stage II or higher bladder cancer, were sequenced, sectioned with hematoxylin-eosin staining, and scanned for WSIs. To facilitate the reuse of these unique datasets, we described the datasets including WSIs and mutational information from whole-exome sequencing(WES) data for bladder cancer in detail with specific attention to data quality. A schematic overview of the study design and workflow is shown in Fig. 1.

A schematic overview of this study design and workflow. PBMCs, peripheral blood mononuclear cells; WSI, whole-slide image.
Methods
Patients and ethical approval
The study was approved by the research ethics committee of the Fudan University Shanghai Cancer Center (FUSCC; approval number 2006218-Exp3). All specimens for 75 patients were obtained retrospectively from the histopathology archive with approval from the FUSCC. The requirement for informed consent was waived by the ethics committee. The clinical information for the 75 patients was shown in Table S1.
Selection and preparation of specimens
Tumor tissues from the 75 patients were routinely fixed in formalin and embedded in paraffin. The tissue sections were stained with hematoxylin-eosin using an automated slide-staining machine (Autostainer XL + TS5015 + CV5030, Leica, Wetzlar, Germany). Case selection was randomized but only specimens with acceptable tissue quality were included. All images were digitized using a linear full-slide scanner (SQS600P, Teksqray, Shenzhen, China) at a resolution of 0.25 micrometers (40x) per pixel. All images were examined by two pathologists working independently and were found to show no evidence of any significant changes in intensity or color.
Pathological assessment and quality control
We implemented a rigorous selection and quality control process to ensure the quality and consistency of pathological assessment. Histology slides were selected based on their quality and the presence of well-preserved tumor tissues. Slides showing significant artifacts and those with poor staining quality were excluded. Tumor areas in the whole-slide images (WSIs) were identified and marked by two pathologists working independently, and included based on the presence of viable tumor cells and minimal contamination by non-tumor tissue. Any discrepancies between the two pathologists were resolved by consensus. Each WSI was then reviewed for image clarity, color consistency, and accurate representation of the tumor area. If any quality issues were detected, the images were rescanned to ensure the highest quality data.
Instrumentation
The WSIs of bladder cancer were acquired using a digital scanner (Leica AT Turbo, Leica) with a 40 × objective lens. The average dimensions of the WSIs were 97389 × 80638 pixels with a physical size of 24.16 × 19.94 mm.
DNA preparation
Fresh tumor tissues and matched peripheral blood mononuclear cells (PBMCs) from 75 patients were obtained from the FUSCC tissue bank. DNA was extracted from the PBMCs using a DNA kit and placed in tubes containing EDTA as an anticoagulant. The tumor tissues were frozen at −80 °C in liquid nitrogen. The collected DNA was extracted again using a DNeasy Blood & Tissue Kit (Qiagen, Venlo, Netherlands).
Whole exome sequencing
DNA staining was performed for tumor tissues and matched PBMCs using an E210 system (Covaris, Woburn, MA,USA), with 100 ng of DNA cut to obtain an average fragment size of 200 base pairs. Next-generation sequencing libraries for tumor gDNA matched to the gDNA for PBMCs were prepared using DNA library kits (Agilent v6, Agilent Technologies, Santa Clara, CA, USA). A fluorometer and a 2100 Bioanalyzer (Agilent Technologies) were used to measure the mass and fragment size of the prepared library. The libraries were loaded onto a Nextseq platform (Illumina, San Diego, CA, USA) for pair-end sequencing with a read length of 150 base pairs.
Data processing and quality control of WES data
Fastp18 corrected the raw sequencing data by quality control, read filtering and base correction. The cleaned data were also aligned to the reference human genome (UCSC hg38) using the Burrows-Wheeler Aligner19. After removal of duplications and local rearrangements by SAMtools20, core single nucleotide variants and insertions and deletions were identified using Mutect221. Next, somatic alterations were obtained after removing germline alterations from a matched blood sample. We fixed mate pairs, marked PCR duplicates and performed the base quality recalibration using a GATK4 Genome Analysis Toolkit22. Variants labeled as “PASS” were kept for downstream analysis. Variations were annotated using the Ensembl Variant Effect Predictor (VEP)23.
Data Records
The TCIA datasets mentioned in the Background & Summary section can be accessed through TCIA (https://doi.org/10.7937/K9/TCIA.2016.8LNG8XDR)24. The datasets for the 75 patients with bladder cancer have been recorded as 112 WSIs files, along with a summary containing mutational information for 16 genes from the WES data (Table S1). The mutational information for the 75 tumor tissues specimens is annotated by VEP containing only PASSed variants. All WSIs are provided as SVS files. Details of the raw sequencing data and matched WSIs are shown in Table S2. These files have been deposited in The National Omics Data Encyclopedia (NODE) (OEP004732)25. The raw sequencing of bladder cancer and germline data have been deposited in the National Genomics Data Center (NGDC) (HRA007156)26.
Technical Validation
Quality control and validation of WSIs
After successfully scanning each slide on the whole slide scanner, a pathologist first independently reviewed all cases in the form of digital slides, and each WSI was reviewed by two pathologists working independently. The pathologists reported all cases according to their routine workflow. The quality and consistency of the slide digitization process was primarily managed and regulated by the WSI quality control module, so as to improve the accuracy and reliability of the digital pathological diagnosis. The quality of scans, sections, diagnosis and data were mainly determined by the pathologists. Figure 2a,c and partially enlarged images (Fig. 2b,d) were independently confirmed to be WSIs of bladder cancer by the two pathologists.

Whole-slide images of tumor tissues from two bladder cancer patients. (a) Whole-slide image of patient 1. (b) Magnified view of the tumor region from patient 1. (c) Whole-slide image of patient 2. (d) Magnified view of the tumor region from patient 2.
Our dataset consisted of 112 WSIs from 75 patients. We counted the distribution of WSIs width and height of the WSIs in pixels. Figure 3 showed that the majority of WSIs had a width in the range of 80,000-90,000 pixels and a height in the range of 90,000-100,000 pixels.

Distribution of width and height of whole-slide images in pixels. WSIs, whole-slide images.
Quality control and evaluation of WES
We used Fastqc27 to assess the quality of the raw data. Fastp was then used to rectify the raw sequencing data by quality control, read filtering, and base correction. Figure 4 showed the Q20/Q30 line charts and the GC content for both the raw and clean data for the 75 samples. The Q20 value (indicating a 99% probability of correct identification) and Q30 value (indicating a 99.9% probability of correct identification) for the clean data surpassed 98% and 94%, respectively, for tumor tissues and 96% and 90% for PBMCs after quality control in most samples. The GC contents remained relatively stable, indicating that no significant errors were introduced during quality control and the consistency of the data was maintained. All data files fell within acceptable parameters.

Quality control of sequencing data. (a–d) The Q20/Q30 of the raw and clean sequencing data. (e,f) The GC content of the raw and clean sequencing data. Each spot or bar on the x-axis represents a single sample. PBMCs, peripheral blood mononuclear cells.
Qualimap28 was used to analyze the sequencing alignment data and assess the features of the mapped reads to identify biases in the sequencing and mapping processes, thereby facilitating decision-making for further analysis. The insert size of bladder cancers and PBMCs were in the range of 184-326 base pairs, resulting in a more uniform read coverage for WES29 (Fig. 5a). The mapping ratio of target reads between tumor tissues and PBMCs was similar, with both hovering around 80% (Fig. 5b). The average depth was calculated to be 146.1 for tumor tissues and 138.6 for PBMCs (Fig. 5c). We also compared the genomic coverage between tumor tissues and PBMCs using coverage depths of 30×, 50×, and 100× as shown in Fig. 5d–f. All the aforementioned data were within the standard range.

Confirmation of sequencing quality and coverage. (a) Insert size distribution of tumor tissues and matched PBMCs. (b) Mapping ratio of target reads for tumor tissues and matched PBMCs. (c) Average depth of coverage for tumor tissues and matched PBMCs. (d–f) Coverage at 30×, 50× and 100× for tumor tissues and matched PBMCs. Each spot or bar on the x-axis represents an individual sample. PBMCs, peripheral blood mononuclear cells.
Statistics and evaluation of variation
We aligned the sequences to the hg38 reference human genome and annotated them using VEP. The variants were predominantly categorized as exonic or intronic, with most exonic variants being nonsynonymous single nucleotide variants, synonymous single nucleotide variants and stop-gain mutations (Fig. 6a). The majority of variants were single nucleotide polymorphisms which accounted for 96.78% of case, while the rest were deletion, double nucleotide polymorphism, insertion, and triple nucleotide polymorphism variants (Fig. 6b). Figure 6c showed the variants observed in a single sample.

Annotation of identified variants. (a) Genomic ___location and the main variant consequences. Classification of variants for all samples (b) and a single sample (c). DEL, deletion; DNP, double nucleotide polymorphism; INS, insertion; SNP, single nucleotide polymorphism; SNV, single nucleotide variant; TNP, triple nucleotide polymorphism; UTR, untranslated region.
We analyzed and counted the top ten mutated genes, including TP53, KMT2D, and KDM6A, in 75 tumor tissues (Table S1). We also documented six other clinically significant genes (Table S1). ERCC2, ERBB2, ATM, and RB1 were associated with the effect of chemotherapy or immunotherapy17. FGFR3 was related to targeted therapy and the prognosis30, while PIK3CA could serve as a potential biomarker for targeted therapy31. Furthermore, we analyzed single nucleotide variants and copy-number variations for all genes. Detailed information on the variations was provided in Tables S3 and S4.
A heatmap illustrating the associations between the mutations of these 16 genes was shown in Fig. 7. Each square in the figure represented the correlation between two genes, with the depth of color indicating the co-occurrence of mutations. Darker colors indicated stronger co-occurrence and lighter colors suggested that mutations in the two genes were more likely to be mutually exclusive. An asterisk indicated a statistically significant correlation, for example, NEB and KMT2D. A dot indicated a tendency toward significance, as with ERBB2 and ERCC2. The results showed that most of these mutations displayed co-occurrence (Fig. 7), indicating that these genes might work together in similar pathways, potentially contributing to the development and progression of bladder cancer.

Heatmap for mutational associations between 16 genes. The color intensity reflects co-occurrence of mutations, with darker colors indicating stronger co-occurrence. Asterisks (*) denote p < 0.05, indicating a highly significant correlation, while a dot (·) represents 0.05 < p < 0.1, indicating a tendency toward significance.
Usage Notes
The dataset for the WSI files described in this article can be downloaded from The National Omics Data Encyclopedia (https://www.biosino.org/node) by pasting the accession number OEP004732 into the text search box or from the URL: https://www.biosino.org/node/project/detail/OEP004732. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA007156) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
The sequencing data were processed by freely available and open access tools. The sequencing data for the matched PBMCs was used to generate a panel of normal in the step of variant detection conducted by Mutect2, confirming that we only provided the mutational information for tumor tissues.
The WSIs can be viewed by ImageScope (https://www.leicabiosystems.com/digital-pathology/manage/aperio-imagescope/). The clinical and mutational information were shown in Table S1. The filenames of the WSIs and raw sequencing data are provided in Table S2. The single nucleotide variants and copy-number variation can be found in Table S3 and Table S4, respectively.
Code availability
We used a typical workflow to process the WES data. All software used in this study is freely available: Fastp: https://github.com/OpenGene/fastp. BWA: https://github.com/lh3/bwa. GATK: https://github.com/broadinstitute/gatk/releases. VEP: https://m.ensembl.org/info/docs/tools/vep/script/vep_download.html. No code was used in the generation of the WSIs files. No code is required to access or analyze this dataset.
References
Lenis, A. T. et al. Bladder Cancer: A Review. JAMA 324, 1980–1991, https://doi.org/10.1001/jama.2020.17598 (2020).
Li, R. Y. et al. Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science 370, 82–89, https://doi.org/10.1126/science.aba7300 (2020).
Hayashi, T. et al. Mutational Landscape and Environmental Effects in Bladder Cancer. Int. J. Mol. Sci 21, 6072, https://doi.org/10.3390/ijms21176072 (2020).
Meng, J. et al. Tumor immune microenvironment-based classifications of bladder cancer for enhancing the response rate of immunotherapy. Mol Ther Oncolytics 20, 410–421, https://doi.org/10.1016/j.omto.2021.02.001 (2021).
Guercio, B. J. et al. Developing Precision Medicine for Bladder Cancer. Hematol Oncol Clin North Am 35, 633–653, https://doi.org/10.1016/j.hoc.2021.02.008 (2021).
Baylin, S. B. & Jones, P. A. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol 8, a019505, https://doi.org/10.1101/cshperspect.a019505 (2016).
Beane, J. et al. Genomic approaches to accelerate cancer interception. Lancet Oncol 18, e494–e502, https://doi.org/10.1016/S1470-2045(17)30373-X (2017).
Brachova, P. et al. TP53 oncomorphic mutations predict resistance to platinum- and taxane-based standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int J Oncol 46, 607–18, https://doi.org/10.3892/ijo.2014.2747 (2015).
Friboulet, L. et al. Molecular Characteristics of ERCC1-Negative versus ERCC1-Positive Tumors in Resected NSCLC. Clin Cancer Res 17, 5562–5572, https://doi.org/10.1158/1078-0432.CCR-11-0790 (2011).
Yang, Z. et al. Somatic FGFR3 Mutations Distinguish a Subgroup of Muscle-Invasive Bladder Cancers with Response to Neoadjuvant Chemotherapy. EBioMedicine 35, 198–203, https://doi.org/10.1016/j.ebiom.2018.06.011 (2018).
Zou, Y. et al. Immune-related gene risk score predicting the effect of immunotherapy and prognosis in bladder cancer patients. Front Genet 13, 1011390, https://doi.org/10.3389/fgene.2022.1011390 (2022).
Wang, F. et al. Comparative Analysis of Differentially Mutated Genes in Non-Muscle and Muscle-Invasive Bladder Cancer in the Chinese Population by Whole Exome Sequencing. Front Genet 13, 831146, https://doi.org/10.3389/fgene.2022.831146 (2022).
Shi, R. et al. APOBEC-mediated mutagenesis is a favorable predictor of prognosis and immunotherapy for bladder cancer patients: evidence from pan-cancer analysis and multiple databases. Theranostics 12, 4181–4199, https://doi.org/10.7150/thno.73235 (2022).
Jang, H. J. et al. Prediction of genetic alterations from gastric cancer histopathology images using a fully automated deep learning approach. World J Gastroenterol 27, 7687–7704, https://doi.org/10.3748/wjg.v27.i44.7687 (2021).
Sarker, I. H. et al. Deep Learning: A Comprehensive Overview on Techniques, Taxonomy, Applications and Research Directions. SN COMPUT. SCI 2, 420, https://doi.org/10.1007/s42979-021-00815-1 (2021).
Clark, K. et al. The Cancer Imaging Archive (TCIA): Maintaining and Operating a Public Information Repository. J Digit Imaging 26, 1045–1057, https://doi.org/10.1007/s10278-013-9622-7 (2013).
Yan, R. et al. Histopathological bladder cancer gene mutation prediction with hierarchical deep multiple-instance learning. Med Image Anal 87, 102824, https://doi.org/10.1016/j.media.2023.102824 (2023).
Chen, S. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008, https://doi.org/10.1093/gigascience/giab008 (2021).
Benjamin, D. et al. Calling Somatic SNVs and Indels with Mutect2. bioRxiv, 861054, https://doi.org/10.1101/861054 (2019).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol 17, 122, https://doi.org/10.1186/s13059-016-0974-4 (2016).
Kirk et al. The Cancer Genome Atlas Urothelial Bladder Carcinoma Collection. The Cancer Imaging Archive https://doi.org/10.7937/K9/TCIA.2016.8LNG8XDR (2016).
Shen et al. NODE https://www.biosino.org/node/project/detail/OEP004732 (2023).
Shen et al. GSA for Human https://ngdc.cncb.ac.cn/gsa-human/browse/HRA007156 (2024).
de Sena Brandine, G. & Smith, A. D. Falco: high-speed FastQC emulation for quality control of sequencing data. F1000Res 8, 1874, https://doi.org/10.12688/f1000research.21142.2 (2019).
García-Alcalde, F. et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678–2679, https://doi.org/10.1093/bioinformatics/bts503 (2012).
Pommerenke, C. et al. Enhanced whole exome sequencing by higher DNA insert lengths. BMC Genomics 17, 399, https://doi.org/10.1186/s12864-016-2698-y (2016).
Loriot, Y. et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med 381, 338–348, https://doi.org/10.1056/NEJMoa1817323 (2019).
Zeng, S. X. et al. The Phosphatidylinositol 3-Kinase Pathway as a Potential Therapeutic Target in Bladder Cancer. Clin Cancer Res 23, 6580–6591, https://doi.org/10.1158/1078-0432.CCR-17-0033 (2017).
Acknowledgements
This research was supported by grants from the National Key Research and Development Program of China (2021YFF1201005), the Informatization Plan of Chinese Academy of Sciences (CAS-WX2021SF-0101), and the Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR4031). The authors thank Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the English text of a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
P.H.X. designed the study and revised the manuscript; T.Q.L., J.W. and H.L.G. collected the tumor tissues and PBMCs and prepared the raw sequencing data and WSIs. F.M.Q. wrote the manuscript; M.K.T. processed the data; D.W.Y., F.R. and Y.J.S. supervised the study. All authors approved the final draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, PH., Li, T., Qu, F. et al. Comprehensive Collection of Whole-Slide Images and Genomic Profiles for Patients with Bladder Cancer. Sci Data 11, 699 (2024). https://doi.org/10.1038/s41597-024-03526-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03526-3
