
Abstract
Endemic to the upper and middle reaches of the Yangtze River in China, elongate loach (Leptobotia elongata) has become a vulnerable species mainly due to overfishing and habitat destruction. Thus far, no genome data of this species are reported. As a result, lacking of such genomic information has restricted practical conservation and utilization of this economic fish. Here, we constructed chromosome-level genome assemblies for both male and female elongate loach by integration of MGI, PacBio HiFi and Hi-C sequencing technologies. Two primary genome assemblies (586-Mb and 589-Mb) were obtained for female and male fishes, respectively. Indeed, 98.22% and 98.61% of the contig sequences were anchored onto 25 chromosomes, with identification of 26.22% and 25.92% repeat contents in both assembled genomes. Meanwhile, a total of 25,215 and 25,253 protein-coding genes were annotated, of which 97.41% and 98.8% could be predicted with functions. Taken together, our genome data presented here provide a valuable genomic resource for in-depth evolutionary and functional research, as well as molecular breeding and conservation of this economic fish species.
Similar content being viewed by others
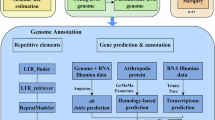

Background & Summary
Elongate loach, Leptobotia elongata, is an endemic freshwater fish species in the upper and middle reaches of the Yangtze River in China1. It is well known for its rapid growth trait and exceptional ornamental value2,3. However, the wild population of this species has significantly declined due to overfishing and habitat destruction4. As a result, this species has been listed as a vulnerable grade (VU) fish in the China Red Data Book of Endangered Animals and the Red List of China’s Vertebrates4,5. Previous studies primarily focused on reproductive and developmental biology6,7,8,9, feeding ecology1, taxonomy, and genetic diversity2,10,11. Meanwhile, the mitogenome and two transcriptomes of this endangered species were also reported3,4,12,13. However, whole genome data are still very limited, which has largely restricted biological research and conservation of this valuable loach species.
In recent years, lots of efforts have been made by Chinese government and scientists to recover and protect wild populations of elongate loach. One of the most important ways is to release artificially bred juvenile loaches into wild rivers and lakes within the upper and middle reaches of the Yangtze River6. In many cases, it is important to know the genders of examined fish; however, it is hard to distinguish the exact sex of loach individuals by morphological observation9. Sex-related specific markers are indeed helpful for sex identification at the early developmental stages14,15, but lacking of genome data has delayed the process to identify such valuable markers. Meanwhile, elongate loach grows rapidly with the biggest body weight and body size in Cobitidae fishes3, but the potential genetic basis is still poorly understood without any supportive genomic data. In addition, our recent work revealed that some species may have experienced an evolution process of genome polyploidization in Cobitidae fishes, including wide-bodied sand loach (Sinibotia reevesae) but not for elongate loach16. More available genome assemblies of Cobitidae fishes will benefit for exploration of such an interesting evolutionary phenomenon. Notably, genome data will also be useful for resolving other scientific issues such as nutrition requirement, breeding, disease control and prevention, as well as species conservation. Thus, a high-quality genome assembly for any target species is often required for both theoretical research and practical protection.
In this study, we obtained two chromosome-level genome assemblies for both male and female elongate loaches by combination of MGI, PacBio, and Hi-C sequencing technologies. Subsequently, the completeness, content, and annotation of both assembled genomes were evaluated in accordance with our previous studies17,18,19. Then, the evolutionary ___location of this loach was reevaluated based on the genome data of twelve representative fishes. Finally, chromosome synteny was compared and analyzed among genomes of elongate loach and other three representative fishes. These genome data presented here provide a valuable genetic resource for future evolutionary and functional research, as well as molecular breeding and conservation of elongate loach for improvement of its economic and ecological values.
Methods
Sample collection
Adult female and male elongate loaches (Fig. 1a,b) were collected from Key Laboratory of Sichuan Province for Fishes Conservation and Utilization in the Upper Reaches of the Yangtze River, Neijiang City, Sichuan Province, China. Muscle tissues from each individual were separately pooled for whole genome sequencing, including MGI short-read, PacBio HiFi long-read, and Hi-C sequencing technologies. Meanwhile, muscle, ovary/testis, brain, skin, spleen, eye, kidney, intestine, gill, heart, stomach, and liver (12 tissues) were collected for transcriptome sequencing (Table 1). These samples were cut into small pieces and freshly frozen in liquid nitrogen, and then were stored at −80 °C before use.

K-mer (17-mer) distribution curves for estimation of the genome size of L. elongata. (a) Female. (b) Male. Photos of the two sequenced individuals are provided for comparison.
The animal experiments were conducted according to the Chinese Ministry of Science and Technology Guiding Directives for Humane Treatment of Laboratory Animals and approved by the Animal Care and Use Committee of Neijiang Normal University.
DNA extraction and genome sequencing
Extraction and purification of genomic DNA (gDNA) from the muscle tissues was carried out using the classic phenol-chloroform method20. Quality and quantity of the extracted DNA were assessed using Nanodrop spectrophotometer and Qubit Fluorometer (Thermo Fisher Scientific, Franklin, MA, USA), respectively.
The gDNA was randomly fragmented to construct a library with an insert-size of 350 bp by using MGIEasy universal DNA library prep set (MGI, Shenzhen, China) for subsequent sequencing on a DNBSEQ T7 platform (MGI). A total of 61.59 Gb and 70.69 Gb of paired-end raw reads (150 bp in length) from female and male were generated, and then they were filtered by SOAPfilter v2.221 (parameter: -i 350 -M 2) to remove low-quality reads and adaptor sequences. Finally, approximately 45.22 Gb (77.96×) and 50.4 Gb (86.89×) of clean reads were obtained (Table 1) for estimation of the genome size and subsequent assembling.
For the PacBio HiFi long-read sequencing, gDNA was used to construct long-read libraries by using a SMRTbell Express Template Prep Kit 2.0 based on PacBio’s standard protocol (Pacific Biosciences, Menlo Park, CA, USA), which were sequenced through a PacBio Sequel II System. A total of 38.41 Gb and 42.52 Gb HiFi reads with N50 sizes of 17,684 bp and 17,435 bp respectively were obtained (Table 1) using the CCS v6.0.022 (Circular Consensus Sequencing) software with the parameter -min-passes 3. These sequencing data covered approximately 66.22× and 73.31× of the female and male genomes, respectively.
For the high-throughput chromosome conformation capture (Hi-C) sequencing, muscle tissues from the female or male individual were collected, and two Hi-C libraries were constructed by using GrandOmics Hi-C kit (the applied restriction enzyme is DpnII; GrandOmics, Wuhan, China) according to the manufacturer’s protocol. The Hi-C libraries were then sequenced using DNBSEQ T7 platform (MGI) with the paired-end module. In total, 99.67 Gb and 102.43 Gb of raw reads were generated for female and male, respectively. Subsequently, fastp v0.19.523 was applied to filter the adaptors and low-quality reads. Finally, high-quality clean reads (81.67 and 83.45 Gb; Table 1) were retained for construction of chromosomes.
RNA extraction and transcriptome sequencing (RNA-seq)
Total RNA was extracted from 12 tissues (Table 1) using a standard Trizol protocol (Invitrogen, Frederick, MD, USA), and purified using a Qiagen RNeasy mini kit (Qiagen, Germantown, MD, USA). RNA with equal amounts from each tissue was mixed for construction of Illumina cDNA libraries followed the manufacture’s guideline, which was then sequenced on a HiSeq X Ten platform (Illumina, San Diego, CA, USA). Around 15.0 Gb of transcriptome data (for each tissue; see more details in Table 1) were generated for assistance to gene annotations.
Genome-size estimation
To estimate the genome size for elongate loach, a k-mer analysis was performed by using MGI short clean reads. Through the k-mer counting (KMC) program and genome character estimator (GCE) v1.0.2 software24, a 17-mer frequency was calculated. The genome size was estimated based on the following formula: G = K_num/K_depth, where G is the genome size, K_depth represents the k-mer depth, and K_num stands for the total number of 17-mers. The genome size for female and male elongate loach were therefore estimated to be about 589 Mb and 595 Mb, respectively (Fig. 1), which are similar to the genome size of Triplophysa dalaica, a closely related plateau fish under the same family Cobitidae25.
De novo genome assembly and chromosome construction
For the initial genome assembly, 38.41 Gb and 42.52 Gb HiFi long reads (Table 1) were de novo assembled into contigs through HiFiasm v0.16.026 with default parameters. The primary female and male genome assemblies were 585 Mb and 588 Mb in length, which are consistent with the estimated genome sizes (Fig. 1), respectively. Based on this primary genome assembly, Hi-C short reads were subsequently employed to construct chromosomes for elongate loach.
First, the Hi-C clean reads were mapped to the assembled contigs using bowtie2 v2.2.527 (--very-sensitive -L 20 --score-min L, -0.6,-0.2 --end-to-end). Subsequently, the HiC-Pro v2.8.128 pipeline was applied to detect valid ligation products and only valid contact paired reads were retained for further analysis. Based on these valid reads, the primary assembly was oriented, ordered, and clustered onto chromosomes using 3D-DNA v3.0 software29 with default parameters. Juicebox v1.11.0830 was employed to visualize before manually adjusting the candidate assemblies. These assemblies had a total length of 586 Mb and 589 Mb, containing 58 and 67 scaffolds with scaffold N50 sizes of 22.85 Mb and 22.62 Mb for female and male, respectively. Finally, a total of 25 chromosomes for each genome were obtained, which contained 98.22% (576 Mb) and 98.61% (581 Mb) of the assembled contigs for female and male, respectively (Fig. 2, Table 2).

Genome-wide analysis of chromatin interactions at 500-kb resolution in (a) female or (b) male genome. Color blocks represent the interactions, with various strength from yellow (low) to red (high).
We employed two methods to evaluate the genome completeness. First, BUSCO v5.0 (Benchmarking Universal Single-Copy Orthologs)31 was employed to search against the actinopterygii_odb10 database. Both female and male assemblies were validated to contain 97.3% [S:96.4%, D:0.9%, F:0.3%, M:2.4%] and 97.6% [S:96.5%, D:1.1%, F:0.3%, M:2.1%] of the 3,640 conserved genes, respectively. Second, based on the BGI short reads, we assessed the consensus quality values (QV) of the two assemblies through Merqury v1.332 with “k-mer = 19”. For female and male assemblies, the mapping rates of reads were 99.58% and 99.63%, respectively (Table 2). These results show that the two genome assemblies have considerable integrity, continuity, and accuracy.
Annotation of repeat elements
Repeat elements (REs) in the elongate loach genome were predicted by combination of homology-based and de novo predictions. For the homology approach, Tandem Repeats Finder v4.0733 was applied to search for tandem repeats. Transposable elements (TEs) were identified using RepeatMasker v4.0.6 and RepeatProteinMask v4.0.634. For the de novo approach, RepeatModeler v1.0.835 and LTR_FINDER v1.0.636 were employed to generate a de novo repeat library, and RepeatMasker was used to annotate REs against this repeat library. A total of 153.81 Mb (26.22%) and 152.72 Mb (25.92%) repetitive sequences were annotated in the female and male genomes (Table 2), in which DNA transposons made up the greatest proportion (10.78% and 10.58%), followed by LTR (10.04% and 9.99%) and LINEs (7.09% and 6.93%, respectively). Compared with the genome of Triplophysa dalaica (REs account for 35.01%), elongate loach displayed comparatively low percentage of REs. Subsequently, the repetitive regions of each genome were masked prior to further gene prediction.
Gene annotation and functional assignment
Prediction of protein-coding genes was carried out using three methods, including homology, ab initio and RNA-seq-based. First, AUGUSTUS v3.2.137 was employed to perform the ab inito gene predictions. Second, GeMoMa v1.6.438 was applied for the homology-based prediction. We aligned homology proteins from six other fish species, including zebrafish (Danio rerio), spotted gar (Lepisosteus oculatus), Indian major carp (Labeo rohita), fathead minnow (Pimephales promelas), Triplophysa bleekeri, and Onychostoma macrolepis (downloaded from the NCBI). Third, the RNA-seq data from twelve tissues were mapped onto the assembled genomes using Trinity v2.5.139, and then gene structures were identified using PASA v2.3.340. Finally, gene sets were integrated by the Evidence Modeler (EVM) pipeline v1.040.
A total of 25,215 and 25,253 protein-coding genes were predicted in the female and male genomes, respectively. Functions of these genes were annotated by BLASTPing the deduced protein sequences against various public databases, including SwissProt, Interpro, NCBI NR, KEGG and Trembl, with an E-value cutoff of <1e−5. A sum of 24,563 (97.42%) and 24,976 (98.8%) genes were successfully annotated in at least one database for female and male, respectively. Meanwhile, the BUSCO completeness values were calculated to be 94.4% and 95.1% of the total predicted genes for female and male, respectively (Table 3). Interestingly, around more than 50 genes were predicted in the male genome assembly, which may be helpful for developing male-specific markers or sex-determining genes for ongoing practices of mono-sex or molecular breeding of this economic fish species.
Synteny analysis
Based on the protein-coding sequences and gene structures, JCVI v19021341 was used to perform a chromosomal synteny analysis among L. elongate, D. rerio, T. bleekeri and T. dalaica. It seems that these fish species have a good collinearity relationship, and their chromosomes present one-to-one correspondence (see Fig. 3), indicating that the assembled genomes are complete and high-quality.

Genome synteny between elongate loach, zebrafish and two Triplophysa loaches.
Data Records
The genome assembly and raw reads of the genome and transcriptome sequencing for elongate loach were deposited at NCBI under the accession number PRJNA108205742. Raw reads are available in the Sequence Reads Archive (SRA) with the accession number SRP49577243. Both genome assemblies were deposited at NCBI GenBank with the accession numbers GCA_039881065.144 and GCA_039881075.145. The annotation files of Leptobotia longiformis are available in Figshare46.
Technical Validation
The quality scores across all bases of the MGI raw sequencing data were inspected using FastQC v0.11.947 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). We conducted a 17-mer distribution analysis to estimate the target genome size based on the MGI clean data. The integrity of assembled genomes and protein-coding genes was evaluated using BUSCO with the actinopterygii_odb10 database. More than 94% of complete BUSCOs were identified in both assembled genomes and annotated protein-coding genes. The synteny analysis of 25 chromosomes between elongate loach, zebrafish and two Triplophysa fishes proved high conservation of synteny between each pair of the four species, indicating that the genome assemblies and annotation for elongate loach are indeed complete and of high quality.
Code availability
The versions and parameters of bioinformatic tools applied in this study have been described in the Method section. If no parameter is provided, the default is set. No custom code was used.
References
Li, L. et al. Diet of Leptobotia elongata revealed by stomach content analysis and inferred from stable isotope signatures. Environ. Biol. Fishes 98, 1965–1978 (2015).
Liu, D. et al. No decline of genetic diversity in elongate loach (Leptobotia elongata) with a tendency to form population structure in the upper Yangtze River. Glob. Ecol. Conserv. 23, e01072 (2020).
Zhang, Y. et al. Transcriptome Sequencing of the Endangered Species Elongate Loach (Leptobotia elongata) From the Yangtze River: De novo Transcriptome Assembly, Annotation, Identification and Validation of EST-SSR Markers. Front. Mar. Sci. 8, 616727 (2021).
Ke, Z. et al. Characterization of the Complete Mitochondrial Genome of the Elongate Loach and Its Phylogenetic Implications in Cobitidae. Animals 13, 3841 (2023).
Jiang, Z. et al. Red List of China’s Vertebrates. Biodivers. Sci. 24, 500–551 (2016).
Liang, Y. et al. Studies on artificial propagation of Leptobotia elongata. Acta Hydrobiol. Sin. 25, 422–424 (2001).
Yin, J. et al. The ovarian cycle of the fish Leptobotia elongata Bleeker, endemic to China. Pak. J. Zool. 44, 997–1005 (2012).
Yang, K. et al. Otolith fluorescent and thermal marking of elongate loach (Leptobotia elongata) at early life stages. Environ. Biol. Fishes 99, 687–695 (2016).
Zheng, Y.-H. et al. Artificial Propagation and Embryonic Development Observation of Leptobotia elongata from Jinsha River and Yangtze River. Hubei Agric. Sci. 57, 104 (2018).
Liu, G. et al. Mitochondrial DNA reveals low population differentiation in elongate loach, Leptobotia elongata (Bleeker): implications for conservation. Environ. Biol. Fishes 93, 393–40 (2012).
Liu, D. et al. High genetic diversity and weak population structure of Leptobotia elongata from different markers. Glob. Ecol. Conserv. 50, e02852 (2024).
Li, P. et al. The complete mitochondrial genome of the Elongate loach Leptobotia elongata (Cypriniformes: Cobitidae). Mitochondrial DNA 23, 352–354 (2012).
Zhang, Y. et al. De novo gonad transcriptome analysis of elongate loach (Leptobotia elongata) provides novel insights into sex-related genes. Comp. Biochem. Physiol. Part D Genomics Proteomics 42, 100962 (2022).
Han, C. et al. Screening and characterization of sex-specific markers developed by a simple NGS method in mandarin fish (Siniperca chuatsi). Aquaculture 527, 735495 (2020).
Mou, C.-Y. et al. Genome-wide association study reveals growth-related markers and candidate genes for selection in Chinese longsnout catfish (Leiocassis longirostris). Aquaculture 560, 738513 (2022).
Lv, Y. et al. Deciphering genome-wide molecular pathways for exogenous Aeromonas hydrophila infection in wide-bodied sand loach (Sinibotia reevesae). Aquac. Rep. 35, 102033 (2024).
Bian, C. et al. A chromosome-level genome assembly for the astaxanthin-producing microalga Haematococcus pluvialis. Sci. Data 10, 511 (2023).
Zhang, K. et al. A chromosome-level reference genome assembly of the Reeve’s moray eel (Gymnothorax reevesii). Sci. Data 10, 501 (2023).
Liu, C. et al. Whole genome sequencing of a novel sea anemone (Actinostola sp.) from a deep-sea hydrothermal vent. Sci. Data 11, 102 (2024).
Köchl, S. et al. DNA extraction and quantitation of forensic samples using the phenol-chloroform method and real-time PCR. Fore. DNA Typ. Protoc. 297, 13–29 (2005).
Li, R. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967 (2009).
Rhoads, A. & Au, K. F. PacBio Sequencing and Its Applications. Genomics Proteomics Bioinformatics 13, 278–289 (2015).
Chen, S. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv preprint arXiv 1308.2012 (2013).
Zhou, C. et al. The Chromosome-Level Genome of Triplophysa dalaica (Cypriniformes: Cobitidae) Provides Insights into Its Survival in Extremely Alkaline Environment. Genome Biol. Evol. 13, evab153 (2021).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Simao, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics Chapter 4, 4–10 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–268 (2007).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–439 (2006).
Keilwagen, J. et al. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol. Biol. 1962, 161–177 (2019).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512 (2013).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Tang, H. et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genomics 15, 312 (2014).
NCBI Bioproject https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1082057 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP495772 (2024).
Zhang, X. NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_039881065.1 (2024).
Zhang, X. NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_039881075.1 (2024).
Zhang, X. Annotation file of Leptobotia elongata from male and female. Figshare https://doi.org/10.6084/m9.figshare.25322026.v1 (2024).
Brown, J. et al. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33(19), 3137–3139 (2017).
Acknowledgements
This work was financially supported by the Natural Science Fund of Sichuan Province of China (no. 2023NSFSC1221), the Project of Sichuan Provincial Department of Science and Technology (no. ZYZFSC22004), the Research Fund from Key Laboratory of Sichuan Province for Fishes Conservation and Utilization in the Upper Reaches of Yangtze River (no. NJTCSC23-3), the Special Project of Performance Incentive Guidance for Research Institution in Chongqing of China (cstc2022jxjl80009), the National Natural Science Funds (no. 32202967), the Natural Science Fund of Hubei Province of China (no. 2022CFB803), and the Cooperation Fund from Sichuan University (no. 2022H013).
Author information
Authors and Affiliations
Contributions
Z.W., Q.S. and X.Z. conceived and designed the study. Z.W., X.W., Y.L., B.Z., C.Z., P.F. and R.L. collected the samples. X.Z., J.C., Z.W., P.P., Y.L. and Y.L. performed data analysis. W.Z., Y.H. and L.Z. conducted experiments for species identification. J.F. provided valuable suggestions. X.Z. and Z.W. wrote the manuscript. Z.W. and Q.S. revised the manuscript. All authors read and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wen, Z., Wei, X., Chen, J. et al. Chromosome-level genome assemblies of vulnerable male and female elongate loach (Leptobotia elongata). Sci Data 11, 924 (2024). https://doi.org/10.1038/s41597-024-03789-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03789-w
This article is cited by
-
A complete telomere-to-telomere chromosome-level genome assembly of X-ray tetra (Pristella maxillaris)
Scientific Data (2025)
-
A chromosome-level genome assembly of critically endangered Ochetobius elongatus
Scientific Data (2024)
