
Abstract
The mud carp (Cirrhinus molitorella) is an important economic farmed fish, mainly distributed in South China and Southeast Asia due to its strong adaptability and high yield. Despite its economic importance, the paucity of genomic information has constrained detailed genetic research and breeding efforts. In this study, we utilized PacBio HiFi long-read sequencing and Hi-C technologies to generate a meticulously assembled chromosome-level genome of the mud carp. This assembly spans 1,033.41 Mb, with an impressive 99.82% distributed across 25 chromosomes. The contig N50 and scaffold N50 are 33.29 Mb and 39.86 Mb, respectively. The completeness of the mud carp genome assembly is highlighted by a BUSCO score of 98.05%. We predict 25,865 protein-coding genes, with a BUSCO score of 96.54%, and functional annotations for 91.83% of these genes. Approximately 52.21% of the genome consists of repeat elements. This high-fidelity genome assembly is a vital resource for advancing molecular breeding, comparative genomics, and evolutionary studies of the mud carp and related species.
Similar content being viewed by others
Background & Summary
The mud carp (Cirrhinus molitorella) is classified within the order Cypriniformes, family Cyprinidae, subfamily Labeoninae, and the genus Cirrhinus1. This species is distributed across southern China and Southeast Asia and is economically significant in southern China, with aquaculture production reaching 72,800 tons in 20212. Mud carp are recognized for their diverse dietary preferences, robust disease resistance, high productivity, and adaptability to various aquatic environments. In South China, it is a prominent species in pond aquaculture and is often listed among the four major economic carp species in Guangdong province, alongside grass carp (Ctenopharyngodon idella), silver carp (Hypophthalmichthys molitrix), and bighead carp (Hypophthalmichthys nobilis)3. Renowned for its tender, flavorful meat at a reasonable price, mud carp enjoys popularity as aquatic products in the market. Additionally, mud carp holds significant value in food processing, with products such as canned mud carp with black beans, mud carp cakes, mud carp balls, and mud carp skin being highly sought after by consumers4. Moreover, mud carp is an important species for leisure fishing in southern China, offering anglers an exhilarating experience with its strong forward rush when hooked, which attracts many fishing enthusiasts.
Genomics is the scientific field focused on studying biological genomes and gene functions5. This field includes sequencing, assembly, functional analysis, and other methodologies, providing crucial insights into the fundamental principles of life6. Recent advancements in second- and third-generation sequencing technologies have greatly improved sequencing efficiency, reduced costs, and expanded the application of these techniques7. Concurrently, genomic research has advanced significantly, particularly in the realm of fish genetics, witnessing a burgeoning number of genomic studies8. Currently, there are over 3,700 species of Cyprinidae distributed across 210 genera worldwide. To date, genome sequencing and assembly have been completed for only a few Cyprinidae species, including Zebrafish (Danio rerio)9, grass carp (Ctenopharyngodon idella)10,11, common carp (Cyprinus carpio)12, goldfish (Carassius auratus)13,14, silver carp (Hypophthalmichthys molitrix)15,16, bighead carp (Hypophthalmichthys nobilis)15,17, Blunt Snout Bream (Megalobrama amblycephala)18, and topmouth culter (Culter alburnus)19. These genome assemblies have facilitated research on species evolution, chromosome rearrangement, and genetic analysis of economic traits, serving as critical foundations for further investigation20,21.
To date, there are no reports on the chromosome-level genome assembly of fishes within the Labeoninae subfamily. Species in the Labeoninae are widely distributed across various water systems south of the Qinling Mountains in China, extending to Southeast Asia and South Asia22. In China alone, there are approximately 40 species and subspecies across 17 genera. Within the Labeoninae subfamily, mud carp holds significant importance in aquaculture. Renowned as the ‘pond scavenger’, mud carp has been cultivated in China for centuries23. As an omnivorous species, mud carp can consume a variety of diets, which facilitating its integration with other fish species to enhance production24. Previous studies on mud carp have primarily focused on its biological and physiological characteristics, growth patterns, dietary habits, population genetics, and sex determination4,25,26. The completion of whole genome sequencing and chromosome-level genome assembly for mud carp will provide a crucial foundation and reference for extensive research, not only on mud carp itself but also on other fish species within the Labeoninae subfamily.
In this study, we utilized PacBio Hifi long-read sequencing and Hi-C technology to generate a high-quality chromosome-level assembly of the mud carp genome. With the development of this high-quality reference genome, we anticipate significant advancements in the field of population genetics and the identification of functional genes associated with key economic traits in mud carp. The elucidation of this genomic foundation holds the potential to pave the way for molecular breeding and gene editing in mud carp.
Methods
Sample collection and DNA extraction
A mature male mud carp was collected from the Pearl River in Guangzhou, China. Muscle tissue from this specimen was utilized for DNA extraction, which was subsequently used for whole-genome sequencing, including short-read, long-read, and Hi-C sequencing. All experiments were conducted following the guidelines set forth by the Ethics Committee of the Pearl River Fisheries Research Institute, Chinese Academy of Fishery Sciences. Genomic DNA was extracted from the muscle tissue using a Qiagen DNeasy Blood and Tissue Kit (Qiagen, USA) according to the manufacturer’s instructions. The quality and concentration of the extracted DNA were evaluated using a NanoDrop One spectrophotometer (Thermo Scientific, USA) and 1% agarose gel electrophoresis.
Genome sequencing
The extracted DNA was randomly sheared into approximately 350 bp fragments, and a short-read library was constructed using the MGIEasy Universal DNA Library Prep Set (MGI, China). Sequencing was performed on the MGISEQ T7 platform (MGI, China), generating a total of 73.91 Gb of paired-end raw reads, each 150 bp in length (Table 1). For PacBio sequencing, a SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, USA) was employed according to PacBio’s standard protocol for HiFi sequencing, and a long-read library was constructed using the PacBio Sequel II system. This process yielded 33.47 Gb of raw PacBio continuous long reads (HiFi) with an average length of 17.13 kb (Table 1). For Hi-C sequencing, approximately 1 g of muscle tissue from the male mud carp was dissected and processed with the GrandOmics Hi-C kit (DpnII restriction enzyme; GrandOmics, China) following the manufacturer’s protocol. The Hi-C library was sequenced on the MGISEQ T7 platform (MGI, China), producing a total of 112.00 Gb of Hi-C read data (Table 1).
RNA extraction and transcriptome sequencing
To facilitate genome annotation, total RNA was extracted from various tissues, including the spleen, kidney, brain, muscle, ovary, and liver. RNA quality was assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). A mixed RNA sample was then used to construct a cDNA library with the MGIEasy Universal DNA Library Prep Set (MGI, China) following the manufacturer’s protocol. This library was sequenced on the MGISEQ T7 platform (MGI, China) with a paired-end 150 bp layout, resulting in 49.63 Gb of transcriptome data to support genome and gene annotations (Table 1).
Genome size and heterozygosity estimation
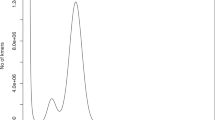
To estimate the genome size of the mud carp, a k-mer analysis was performed using MGI clean reads. Initially, Jellyfish (v2.3.0)27 was used to calculate the frequency of 17-mers and generate a k-mer frequency table. Subsequently, GenomeScope (v2.0)28 was used to analyse the 17-mer frequency table, resulting in an estimated genome size of 884,402,008 bp, with 0.461% heterozygosity and 46.4% unique sequences (Fig. 1).

The 17-mer frequency distribution analysis chart for the Cirrhinus molitorella genome.
Genome assembly
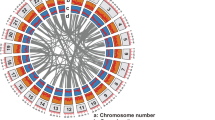
Genome assembly was performed using the default parameters of Hifiasm (v0.16.1)29. HiFi long reads were used as input for Hifiasm to construct primary assembly contig graphs. Overlaps were precomputed and then recalculated from the corrected reads. Haploid duplications were removed using Hifiasm, followed by three rounds of error correction. The assembly yielded 69 contigs with a total length of 1033.41 Mb. The maximum contig size was 54.85 Mb, with an N50 of 33.29 Mb (Table 2). Scaffolding was performed using Juicer (v1.6)30 combined with 3D-DNA (v201008)31. Initially, BWA (v0.7.17)32 was used to index the contig-level genome, followed by Juicer to create restriction enzyme cutting sites. Juicer was used to map clean Hi-C (paired-end) reads to the contigs, and Hi-C-assisted initial chromosome assembly was conducted using the 3D-DNA algorithm following standard procedures. Chromosome boundaries were adjusted, and scaffolds corrected using the manually operated Juicerbox (v1.11.08)33 module, resulting in the resolution of 25 chromosomes (Figs. 2, 3). The modified file output by Juicebox was revised and used as input for 3D-DNA to perform re-scaffolding on a per-chromosome basis. The final assembly consisted of 42 scaffolds, with a maximum scaffold size of 67.41 Mb and an N50 size of 39.86 Mb (Tables 2, 3).

Hi-C contact map produced by 3D-DNA.

Features of the Cirrhinus molitorella genome. From outside to inside: (I) the 25 pseudo-chromosomes, (II) gene density, (III) repeats, (IV) GC content, (V) variant density and (VI) collinear regions (each line connects paralogous genes on diferent chromosomes).
Repeat annotation
In recognition of the significance of tandem repeats, we utilized two software tools, GMATA (v2.2.1)34 and Tandem Repeats Finder (TRF, v4.10.0)35, to conduct a genome-wide search for tandem repeat sequences with default parameters. GMATA primarily identifies SSRs with shorter repeat units, while TRF explores tandem repeats encompassing all types of repeat units. The results indicate that SSRs constitute 0.19% of the total genome length, while tandem repeat sequences account for1.06% of the genome length. Subsequently, we scrutinized the dispersed repetitive sequences. First, we used MITE-hunter36 to identify miniature inverted-repeat transposable elements (MITEs) within the genome, constructing a MITE library file. Subsequently, a hard-masking operation was executed on the genome, marking repeated sequences as ‘N’, and RepeatModeler (v2.05)37 was then used to perform a de novo search for additional repeated sequences, generating a de novo library file (RepMod.lib). Considering that RepMod.lib contains many Unknown repeated sequences, TEclass38 was used for classification. Finally, the MITE.lib, RepMod.lib and Repbase (v19.06)39 libraries were combined to create a comprehensive library file. This combined library file was then employed with RepeatMasker (v4.1.6)40 to search for repeated sequences throughout the entire genome. The results show that dispersed repetitive sequences make up 45.18% of the genome length (Table 4). Among transposable elements (TEs), DNA elements are the most prevalent, comprising 29.37% of the genome, followed by long terminal repeat (LTR) retrotransposons at 5.55%, long interspersed nuclear elements (LINEs) at 4.34%, and short interspersed nuclear elements (SINEs) at 0.55%. In total, 539,498,149 base pairs of repetitive sequences were identified, representing 52.21% of the entire genome (Table 4).
Gene prediction and function assignment
Gene structure prediction was conducted through three distinct methodologies: homology-based, transcriptome-based, and ab initio annotations. For homology-based prediction, we employed GEMOMA (v1.6.1)41 to compare homologous proteins from five related species (Danio rerio, Carassius auratus, Cyprinus carpio, Megalobrama amblycephala, Sinocyclocheilus grahami) with our assembled genome. Transcriptome sequence annotation was conducted using PASA (v2.3.3)42, which provided gene information for semi-supervised self-training of gene models via GeneMark-ST (v5.1)43. The predicted genes were then compared with the SwissProt database44 using BLASTP, with alignments filtered for identity ( ≥ 95%). We selected the top 3,000 genes with the highest alignment scores from GeneMark-ST as the training set for AUGUSTUS model training. AUGUSTUS (v3.5.0)45 was subsequently employed to predict genes within the genome using the trained model. The gene prediction results from ab initio, homology-based, and transcriptome-based methods were converted into a file format compatible with EVM (v2.1.0)42. These files were integrated using EVM with default parameters, producing an initial non-redundant gene set. Our predictions identified a total of 25,865 genes in the genome, with an average gene length of 20,822.68 bp, an average coding sequence length of 1,675.78 bp, and an average of 10.08 exons per gene (Table 5, Fig. 4).

Venn diagram of function annotations from various databases.
Data Records
The raw sequencing reads of all libraries have been deposited into NCBI SRA database via the accession number PRJNA100116446. The assembled genome has been deposited at Genbank under the accession number GCA_033026305.147. Moreover, data of the genome annotations, predicted coding sequences and protein sequences are available at Figshare48.
Technical Validation
Assessment of genome assembly
The accuracy of the mud carp genome assembly was evaluated by assessing its completeness using the conserved metazoan gene set ‘actinopterygii_odb10’ from BUSCO (v5.4.3)49. The analysis demonstrated high completeness, with an overall completeness of 98.1%. Specifically, 96.8% of the genes were complete and single-copy, 1.3% were complete and duplicated, 0.9% were fragmented, and 1.0% were missing. These findings indicate the high quality of the mud carp genome assembly (Table 6).
Gene annotation validation
To evaluate the integrity of the annotated gene set, we conducted BUSCO analysis using conserved single-copy homologous genes from the ‘actinopterygii_odb10’ library. The results revealed that approximately 96.54% of the complete gene elements are present in the annotated gene set, indicating a high level of completeness in the conserved gene predictions. Specifically, 95.08% of the genes were complete and single-copy BUSCOs, with only 0.47% fragmented and 2.99% missing from the assembly (Table 7). These findings highlight the exceptional integrity and conservation of gene content in the dace genome assembly, leading to highly confident prediction outcomes.
Code availability
No special codes or scripts were used in this work, and Data processing was carried out based on the protocols and manuals of the corresponding bioinformatics software. The version and parameters of software have been described in Methods.
References
Yang, C., Zhu, X. & Sun, X. Development of microsatellite markers and their utilization in genetic diversity analysis of cultivated and wild populations of the mud carp (Cirrhina molitorella). Journal of Genetics and Genomics 35, 201–206 (2008).
Wang, Y., Zhao, J., Li, W., Zhang, X. & Zhu, X. Development and characterization of a multiplex microsatellite panel for the mud carp (Cirrhinus molitorella). Conserv Genet Resour 7, 575–577 (2015).
Yu, F. D. et al. The current distribution of invasive mrigal carp (Cirrhinus mrigala) in Southern China, and its potential impacts on native mud carp (Cirrhinus molitorella) populations. Journal of Freshwater Ecology 34, 603–616 (2019).
Liu, H. et al. Sex-specific markers developed by 2b-RAD and genome sequencing reveal an XX/XY sex-determination system in mud carp (Cirrhinus molitorella). Aquaculture 565, 739131 (2023).
Collins, F. S., Green, E. D., Guttmacher, A. E., Guyer, M. S. & Institute, U. S. N. H. G. R. A vision for the future of genomics research. Nature 422, 835–847 (2003).
Brown, T. A. Genomes 5 (CRC Press, 2023).
Robledo, D., Palaiokostas, C., Bargelloni, L., Martínez, P. & Houston, R. Applications of genotyping by sequencing in aquaculture breeding and genetics. Reviews in aquaculture 10, 670–682 (2018).
Fan, G. et al. Initial data release and announcement of the 10,000 Fish Genomes Project (Fish10K). GigaScience 9, giaa080 (2020).
Howe, K. et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503 (2013).
Wang, Y. et al. The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nat Genet 47, 625–631 (2015).
Wu, C.-S. et al. Chromosome-level genome assembly of grass carp (Ctenopharyngodon idella) provides insights into its genome evolution. Bmc Genomics 23, 271 (2022).
Xu, P. et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat Genet 46, 1212–1219 (2014).
Chen, D. et al. The evolutionary origin and domestication history of goldfish (Carassius auratus). Proceedings of the National Academy of Sciences 117, 29775–29785 (2020).
Chen, Z. et al. De novo assembly of the goldfish (Carassius auratus) genome and the evolution of genes after whole-genome duplication. Science advances 5, eaav0547 (2019).
Jian, J. et al. Whole genome sequencing of silver carp (Hypophthalmichthys molitrix) and bighead carp (Hypophthalmichthys nobilis) provide novel insights into their evolution and speciation. Mol Ecol Resour 21, 912–923 (2021).
Zhou, Y., Qin, W., Zhong, H., Zhang, H. & Zhou, L. Chromosome-level assembly of the Hypophthalmichthys molitrix (Cypriniformes: Cyprinidae) genome provides insights into its ecological adaptation. Genomics 113, 2944–2952 (2021).
Fu, B., Zhou, Y., Liu, H., Yu, X. & Tong, J. Updated genome assembly of bighead carp (Hypophthalmichthys nobilis) and its differences between male and female on genomic, transcriptomic, and methylation level. Frontiers in Genetics 12, 728177 (2021).
Liu, H. et al. A chromosome-level assembly of blunt snout bream (Megalobrama amblycephala) genome reveals an expansion of olfactory receptor genes in freshwater fish. Mol Biol Evol 38, 4238–4251 (2021).
Jiang, H. et al. Chromosome‐level genome assembly and whole‐genome resequencing of topmouth culter (Culter alburnus) provide insights into the intraspecific variation of its semi‐buoyant and adhesive eggs. Mol Ecol Resour 23, 1841–1852 (2023).
Li, J.-T. et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat Genet 53, 1493–1503 (2021).
Wang, Y. et al. Comparative genome anatomy reveals evolutionary insights into a unique amphitriploid fish. Nature ecology & evolution 6, 1354–1366 (2022).
Yang, L. & Mayden, R. L. Phylogenetic relationships, subdivision, and biogeography of the cyprinid tribe Labeonini (sensu) (Teleostei: Cypriniformes), with comments on the implications of lips and associated structures in the labeonin classification. Mol Phylogenet Evol 54, 254–265 (2010).
Nguyen, T. T. T. & Sunnucks, P. Strong population genetic structure and its management implications in the mud carp Cirrhinus molitorella, an indigenous freshwater species subject to an aquaculture and culture‐based fishery. J Fish Biol 80, 651–668 (2012).
Bowen, S. H., Gu, B. & Huang, Z. Diet and digestion in Chinese mud carp Cirrhinus molitorella compared with other ilyophagous fishes. Transactions of the American Fisheries Society 135, 1383–1388 (2006).
Caiyan, Z. H. U. et al. Germplasm characteristics and genetic diversity analysis of an original species population of mud carp (Cirrhinus molitorella) from Guangdong Province. South China Fisheries Science 1, 1–5 (2005).
Zhang, D.-C., Huang, Y.-Q., Shao, Y.-Q. & Jiang, S.-G. Molecular cloning, recombinant expression, and growth–promoting effect of mud carp (Cirrhinus molitorella) insulin-like growth factor-I. General and comparative endocrinology 148, 203–212 (2006).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics 25, 1754–1760 (2009).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101 (2016).
Wang, X. & Wang, L. GMATA: an integrated software package for genome-scale SSR mining, marker development and viewing. Frontiers in plant science 7, 215951 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res 38, e199–e199 (2010).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and genome research 110, 462–467 (2005).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 25, 4–10 (2009).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: homology-based gene prediction utilizing intron position conservation and RNA-seq data. Gene prediction: Methods and protocols 1962, 161–177 (2019).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome biology 9, 1–22 (2008).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res 43, e78–e78 (2015).
Bairoch, A. et al. The universal protein resource (UniProt). Nucleic Acids Res 33, D154–D159 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–W439 (2006).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP498955 (2024).
Liu, H. Cirrhinus molitorella isolate prfri, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc.gca:GCA_033026305.1 (2024).
Liu, H. A chromosome-level genome assembly of the mud carp (Cirrhinus molitorella). Figshare https://doi.org/10.6084/m9.figshare.25835875 (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This work is supported by China Agriculture Research System (CARS-46); China-ASEAN Maritime Cooperation Fund (CAMC-2018F); Central Public-interest Scientific Institution Basal Research Fund, CAFS (2023TD37); Guangdong Province Rural Revitalization Strategy Special Fund (2022-SPY-00-016); National Freshwater Genetic Resource Center (FGRC18537); Guangdong Rural Revitalization Strategy Special Provincial Organization and Implementation Project Funds (2022-SBH-00-001), Natural Science Foundation of Jiangsu Province (BK20211367).
Author information
Authors and Affiliations
Contributions
J.Z. and M.O. conceived and designed the study, H.L., T.C. and J.Z. contribute to the materials of this study, H.L. and T.C. collected and analysed the data, H.L. and T.C. contribute to the genome assembly and annotation and uploaded the data, H.L. and T.C. wrote the manuscript, B.L. and L.F. provided critical revision on the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, H., Cui, T., Liu, H. et al. Chromosome-level genome assembly of the mud carp (Cirrhinus molitorella) using PacBio HiFi and Hi-C sequencing. Sci Data 11, 1249 (2024). https://doi.org/10.1038/s41597-024-04075-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-04075-5
