
Abstract
Differences in prognostic outcomes are prevalent in patients with colorectal cancer liver metastases. Comparative analysis of tissue samples, particularly applying single-cell transcriptome sequencing technology, can provide a deeper understanding of potential impacting factors. However, long-term monitoring for prognosis determination necessitates extended preservation of tissue samples using formalin-fixed and paraffin-embedded (FFPE) treatments, which can cause substantial RNA degradation, presenting challenges to single-cell or single-nucleus sequencing. In this study, employing snRandom-seq, a single-nucleus RNA sequencing (snRNA-seq) technology specifically for FFPE samples, we tested multiple lesion samples from 18 distinctive colorectal cancer liver metastasis cases with diverse prognostic outcomes that have been preserved for at least three years (mostly over five years). The process yielded expression data from 82,285 cells. The high-quality snRNA-seq data demonstrate the feasibility of single-nucleus sequencing in long-term preserved FFPE samples, offering potential insights into the heterogeneity between different prognoses of colorectal cancer liver metastases, and the relationship between the heterogeneity within different lesions of the same patient and prognosis.
Similar content being viewed by others


Background & Summary
Colorectal cancer, being the third most common type of malignant tumor globally, and the second leading cause of cancer-related deaths worldwide, has always attracted widespread attention in the medical and scientific research fields. However, when it spreads to the liver, the survival rate of most patients worldwide significantly decreases, while the quality of life is devastatingly impacted, including distressing symptoms, ongoing physical and mental stress, as well as high financial burden1,2. It is perplexing for clinicians and researchers that although all patients with colorectal cancer face the same challenge, there is a vast variability in their clinical prognosis. The factors involve a series of variables such as age, gender, lifestyle, stage of the disease, the patient’s own health status, the origin of the disease, the biological characteristics of the tumor, and the patient’s treatment methods and resistance to disease, among other variables3. At the same time, some past and present research conclusions make us believe that tumor heterogeneity may be the key element that determines the variability in prognosis4. Tumor heterogeneity refers to the differences in cell behavior and characteristics at various locations in the same individual over time and space, including differences in gene expression, metabolic activity, cell vitality and proliferation rate, migration ability, and sensitivity to drugs and other treatment measures5,6. Thorough research into the relationship between the biological properties of colorectal cancer complicated by liver metastasis, tumor heterogeneity and patient prognosis will undoubtedly unveil new knowledge domains, and hold significant scientific and clinical value for optimizing treatment strategies, improving the quality of care, and enhancing patient prognosis7.
In light of this high degree of complexity, researchers need a deep understanding and mastery of its impact on patient prognosis and the relevant mechanisms. Traditional methods such as batch-based FISH and bulk RNA-seq can no longer meet our research needs for the heterogeneity of complex diseases. With the advent and widespread application of single-cell transcriptome sequencing technology without specific mutation requirements, science has undoubtedly provided us with a unique and powerful research tool that can be used to study cell expression profiles in detail, and even find differences in gene expression within the same tumor8.However, when faced with the challenges of sampling, storage methods, and sample quality, we also need to design innovative strategies and means to cope. Conventional clinical tissue samples are usually stored by formalin-fixed and paraffin-embedded (FFPE) for long-term preservation. However, this method may lead to RNA (including mRNA) degradation, which limits its application in RNA sequencing (including single-cell transcriptome sequencing)9,10. When we consider using long-term preserved FFPE samples for research, even for several years, we need to overcome RNA degradation, RNADNA cross-linking issues, and conduct appropriate sample preparation and post processing11.
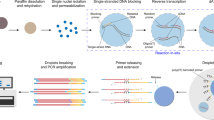
In this study, we have collected 18 FFPE patient samples from different liver metastasis lesions, which have been preserved for at least three years, employing strict inclusion criteria. The criteria details are referred to in the Methods section, and the detailed patient information is available in Table 1. Multiple tumor samples from the same patient were obtained from different lesions. Among these samples, two patients (GP1; GP2) demonstrated a favorable prognosis, with a total of 8 samples collected, and their overall survival exceeded five years. In contrast, three patients (PP1; PP2; PP3) exhibited a poor prognosis, with a total of 10 samples, and their survival duration did not surpass three months. This stark contrast in survival times underscores the significant differences in disease progression and outcomes associated with these patient groups. We performed single-nucleus transcriptome sequencing combining with our previously developed snRandom-seq technology suitable for FFPE samples12. The snRandom-seq can capture total RNAs with random primers (Fig. 1A). Although long-term storage of FFPE samples results in severe degradation, leading to reduced RNA quality and fragmentation, rendering them unsuitable for transcriptome sequencing13, the median number of genes in each sample, except for sample PP1_1, still exceeds 200, with a peak value reaching nearly 800. The count median is around 500. Additionally, the mitochondrial proportion in samples, with a few exceptions, remains relatively low (Fig. 1B). Further annotation analysis based on the marker genes included in the PanglaoDB database14 reveals that, aside from the PP1_1 sample, the remaining samples cover the major cell types of liver tissue, with Hepatocytes and T cells being the predominant cell types (Fig. 1C). snRandom-seq technology provides more applications of single-nucleus transcriptome sequencing on FFPE samples, even for samples with longer preservation periods. More importantly, the data by this study provide an in-depth and more accurate basis for research on colorectal cancer with liver metastasis, potentially suggesting innovative strategies to enhance patient prognosis.

Study design and single nuclei RNA profiling from metastatic colorectal cancer FFPE samples. (A) Diagrammatic illustration of the overall study design. (B) Violin plots illustrate the distribution of feature counts and the proportion of mitochondrial reads for each sample. (C) UMAP plots present the clustering and annotation results for each sample.
Methods
Sample selection and sampling
The samples for this study are derived from colorectal cancer patients with liver metastasis, who underwent surgery at the First Affiliated Hospital of Zhejiang University between 2016 and 2021. All materials collected were FFPE samples. This study has received approval from the Clinical Research Ethics Committee of the First Affiliated Hospital of Zhejiang University School of Medicine (No. IIT20220893A). Considering the retrospective nature of the study, the requirement for informed consent has been waived. This exemption, granted by the committee, acknowledges that the risk associated with data collection is minimal and strictly limited to previously collected medical samples. The ethics committee also approved the release of the data for publication. Sample selection and sampling is orchestrated predicated on several thoughtful principles. Initially, patients harboring unresectable metastases (meaning the number of metastatic lesions is greater than 5) are subjected to transformative treatment. If the transformative treatment proves successful, surgical interventions are planned. It is an obligatory requirement that all lesions get surgically cleared. Subsequently, patients are classified based upon their recurrence pattern and survival period after surgery. This involves segregating patients who suffered short-term recurrence (within one year) and consequently short survival time, from those who did not have an immediate recurrence (greater than three years) and hence demonstrate a longer survival time.
Single nucleus isolation and library preparation
Undergoing detailed preparation for single nucleus isolation and subsequent snRandom-seq library preparation, the surgical samples were scrupulously managed. Initially, five 20 μm sections were incised from each paraffin-embedded surgical sample. To expel paraffin, the samples underwent room temperature xylene washes, usually twice, for a span of five minutes each.
Post paraffin removal, the samples were mildly desiccated using a graded ethanol series, titrated from complete purity to 30%. Subsequently, a pair of washes were administered with a precooled wash buffer, incorporating 125 µm glycine and 2 mM MgSO4 in 3X SSC buffer. Followed by this, the homogenization process was undertaken on an ice-bathed Dounce homogenizer. This step was individualized per sample type, incorporating select lysis buffers and lysis times as necessitated. The homogenizer was then rinsed with a milliliter of lysis buffer. The next move involved the addition of 100 µL protease K (concentration of 10 mg/mL) to the lysis buffer, with a subsequent 5-minute incubation period at 37 °C. The released nuclei were sieved through a stringent 20 µm cell strainer followed by a duo of wash buffer cleansing. The nuclear samples were then parceled equally, DAPI- (4′,6-diamidino-2-phenylindole) stained, then loaded onto a blood cell counter for inspection under an inverted fluorescence microscope. The critical sequence of snRandom-seq library preparation came next. Single nuclei that qualified were processed in line with the meticulous snRandom-seq protocol illustrated in the prior study by Xu12. This comprehensive protocol, inclusive of the nuances like the volumes of lysis buffer, details on permeabilization buffer, and the exacting reaction system and programme, is expanded in the supplementary data of the precedent publication.
Preprocessing of snRandom-seq data
Primarily, raw sequencing data underwent a process to trim off primer sequences and extra nucleotides that were the byproduct of the dA-tailing phase. Following this, for every Read1 instance, an extraction of the UMI (8 nt) and cell-specific barcode (30 nt) was performed, proceeding to merge the organized barcodes. These were then uniquely allotted to the identical acceptor barcode, adhering to a Hamming distance not exceeding 2 nt. Read2 was translated into a gene expression matrix utilizing the STARsolo module nested within STAR (2.7.10a)15, with appropriate parameters (key parameters include:--soloType CB_UMI_Simple --soloCBwhitelist None --soloCBstart 1 --soloCBlen 15 --soloUMIstart 16 --soloUMIlen 8 --outSAMtype BAM SortedByCoordinate --outMultimapperOrder Random --runRNGseed 1 --outSAMattributes NH HI AS nM CB UB GX GN --soloFeatures Gene GeneFull --soloUMIdedup Exact --outSAMunmapped Within --soloStrand Reverse). To elucidate the count of nuclei per sample, a scatterplot of log10(genes) was plotted against each plausible barcode. Here, the minimum peak value of the maximum log10(genes) was adopted as the threshold. Consequently, for downstream analysis, only those barcodes surpassing this gene count threshold were selected. The reference genome used in this study was the human genome version hg38. The genome file was downloaded from the following link: https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_43/GRCh38.primary_assembly.genome.fa.gz. The corresponding genome annotation file was also obtained from: https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_43/gencode.v43.primary_assembly.annotation.gtf.gz.
Clustering and cell annotation
In the investigation of single-nucleus RNA sequencing (snRNA-seq) data, the Seurat v4 toolkit was pivotal for analysis and visual representation16. The process entailed preprocessing, amalgamation, display, congregation, cell type recognition, and detection of differential expression. The study excluded nuclei with gene representation of less than 100 and genes observed in fewer than 3 nuclei. Whether conducting clustering analysis on individual samples or integrating snRNA-seq datasets, the Seurat toolkit was utilized for data preprocessing. Subsequently, the Liger method17, employing non-negative matrix factorization (NMF), was introduced to perform dimensionality reduction on the high-dimensional transcriptome expression matrix. The clustering analysis was then based on this dimensionality reduction result. In detial, Seurat’s embedded functions like NormalizeData, FindVariableFeatures, and ScaleData were executed in succession for preprocessing of the data. Consequently, Liger was applied for integration, using RunOptimizeALS (key parameters include: k = 20, lambda = 5, split.by = “orig.ident”) for dimensionality reduction and RunQuantileNorm (key parameters include: split.by = “orig.ident”) functions to ensure comprehensive integration. Subsequently, clustering analysis was conducted based on the principal components computed through Liger. This process was chiefly executed by the functions FindNeighbors (key parameters include: reduction = “iNMF”, dims = 1:20) and FindClusters (key parameters resolution = 0.3). Clustering outcomes were graphically represented using uniform manifold approximation and projection (UMAP), a feature embedded in Seurat. Stereotypical markers were used to discern the cellular identity of each cluster, manually determined using established marker gene lists. The detection of primary marker genes was accomplished utilizing the Seurat function ‘FindAllMarkers’, enforced with filtering parameters (only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25) to assure uniformity across the study. Furthermore, for the annotated clusters of cell types identified, we utilized marker genes provided in the Panglaodb database14 and manually defined their cell types through the FeaturePlot function. To validate the accuracy of the identified marker genes, we utilized COMETSC18 to construct two-marker gene panels, with all parameters set to their default values. More detailed code has been uploaded to the GitHub repository, and the link can be found in the ‘Code availability’ section.
Identification of lncRNA-mRNA pairs
The LncPairs algorithm19 was used to identify the lncRNA(long non-coding RNA)-mRNA pairs utilized. It began by constructing a gene×cluster expression matrix that revolved around the top 2,000 variations within the gene-based single-cell expression matrix, subsequently averaging the expression of each gene by clusters. This gene×cluster expression matrix was later divided into two separate ones—namely, mRNAcluster and lncRNAcluster. An important part was to calculate the correlation between these two matrices, with lncRNA-mRNA pairs exhibiting a Pearson Correlation Coefficient (PCC) of more than 0.85 being given particular importance. The remaining lncRNA-mRNA pairs were then used in building the paircluster matrix. The identification of cluster-specific lncRNA-mRNA pairs followed, done through the Cosine similarity approach. Finally, pairs that scored below 0.95 in similarity were removed from the dataset.
CNV analysis
Hepatocyte subgroup data was extracted from single-cell transcriptome data to analyze copy number variations (CNVs), with cells from normal samples serving as controls. Utilizing CopyKAT V1.1.020, a Bayesian segmentation approach, each cell was categorized as normal or tumor based on the genome-wide copy number profiles generated from the gene expression Uniquely Mappable Identifier (UMI) matrix. Aneuploid cells that displayed genome-wide copy number aberrations were identified as cancerous, whereas diploid cells were classified as normal cells.
Enrichment analysis
Every gene identified as exhibiting differential expression, even those enriched within particular clusters, proceeded to pathway enrichment analysis, facilitated by the clusterProfiler21. Biological processes were annotated to those pathways, which demonstrated substantial statistical significance.
Data Records
The scRNA-seq data used in this study are accessible through the CNGB Sequence Archive (CNSA) of the China National GeneBank DataBase (CNGBdb) under the BioProject accession PRJCA026536 and are available under the GSA-human Data Usage Agreement22. Users can request access through the CNSA platform by adhering to the controlled access guidelines and completing the GSA-Human Data Access Agreement. The specific access link is https://ngdc.cncb.ac.cn/gsa-human/browse/HRA007565. The final gene expression profiling for each sample had been deposited in FigShare https://doi.org/10.6084/m9.figshare.2592874323.
Technical Validation
Quality assessment of sequencing data
Through an analysis of key sequencing metrics, such as true BC1(%), true BC2(%), valid barcode(%), reads to align(%), median reads, UMI, and genes per cell, we uncovered the inherent complexity and defining characteristics of the dataset. This evaluation covered 18 samples from both tumor and normal tissues (Table 2), providing a robust foundation for further biological interpretation. Initially, we noted a range of sequencing volumes, from 59.7 G to 129.2 G, with the sample PP3_2 featuring the highest sequencing volume at 129.2 G. The raw reads varied between 199 M and 430.8 M. Additionally, in terms of the proportions of real barcodes, we found that the percentages of the three barcodes (True BC1, BC2, BC3) fell within the ranges of 92.8%–97.6%, 90.5%–95.7%, and 88.8%–94% respectively. Moreover, the percentage of valid barcodes ranged from 88% to 93.6%. When considering the alignment of reads, the majority of samples exhibited consistently high proportions (Reads to align) and quantities (Reads to align in million). Most proportion values were around 80%, with the highest alignment quantity reaching 356.8 M. However, sample PP1_1 showed lower proportions (approximately 22.6%) and quantities (around 55.3 M), which we believe may be attributed to the quality of the sample from Patient3. Furthermore, the estimated number of cells ranged from 1828 to 14377, with median reads per single cell ranging from 609 to 7416, median UMI ranging from 314 to 1192, and median gene count extending from 169 to 792. Furthermore, the comprehensive analysis revealed the identification of approximately 18,000 protein-coding genes in each sample, alongside the detection of various non-coding genes such as lncRNA, miRNA, snRNA, snoRNA, and scaRNA. Among these, lncRNA were found to be the most abundant. While there were some discrepancies in the detection quantities across the samples, the majority showed quantities exceeding 10,000, with a peak reaching 15,000, except for samples PP1_1 and PP2_4 (Fig. 2). These indicators effectively demonstrate the depth and accuracy of our sequencing data.

Stacked bar chart illustrating the cell types and corresponding numbers of genes involved in each sample. Each color represents one sample.
Sample integration and cell type annotation
Subsequently, after data integration, we generated a snRNA-seq dataset comprising 82,285 cells. We reannotated these cells into seven major cell types, representing the predominant cell types in liver tissue, with hepatocytes and T cells being the most abundant, totaling 22,008 and 38,701 cells, respectively (Fig. 3A). Specifically, our research has uncovered that Cholangiocytes show distinct expressivity in the KRT7, HNF1B, and CFTR genes; Stellate cells in the COL1A2, COL3A1, and SPARC genes; Endothelial cells in the VWF, CD93, and EMCN genes; Plasma cells in the PIM2, MZB1, and IGHG1 genes; Hepatocytes in the ALB, SERPINA6, and NR1I3 genes; T cells in the CD3E, IL7R, and LEF1 genes; and Kuppffer cells in the CD163, TIMD4, and VSIG4 genes (refer to Fig. 3B). Each sample contained all identified cell types, though their proportions varied (Fig. 3C), demonstrating the experimental success and reliability of the dataset. Furthermore, through the CNV analysis, potential tumor cells can be effectively identified within the hepatocytes group. These cells exhibit significant segmental amplifications and deletions in their genome (Fig. 3D). These findings enhance our comprehension of gene expression patterns in specific cell types within liver tissue and underscore the potential of the snRandom-seq technique for identifying and distinguishing different cell types in long-term preserved, FFPE samples.

Identification of cell types and tumor cells. (A) UMAP plot displays the integrated results of all samples, with all cells categorized into seven major cell types. (B) UMAP plot for the specific expression of three marker genes in different cell types. (C) The proportion of each major cell type across 18 samples, accompanied by the clinical characteristics of each sample as shown. (D) UMAP visualization of the CNV-predicted results, where aneuploid denotes tumor cells, and diploid indicates normal cells. The heatmap of the CNV variations on chromosomes 1–22 and X. Red represents gain, while blue denotes loss.
Identification of cell-type specific gene and lncRNA markers
Identification of cell type-specific genes and lncRNA markers not only provides insights into cell function and changes under disease conditions, but also further confirms the reliability of the data. From the heatmap below, it is clear that each type of cell can be identified by its specific genes and lncRNA markers (Fig. 4A, specific lists can also be found in Table S1, S2). Some of the identified marker genes have been confirmed to correspond to specific cell types, such as PIM2, which is annotated as a gene for B/Plasma cells and also recognized as a marker gene. In the Panglaodb database, CUX2 is also identified as a marker gene for hepatocytes, indicating the accuracy of the data. While the relationship between lncRNA markers and corresponding cell types has not been extensively studied, some lncRNAs have been implicated in colorectal cancer metastasis. For example, the LINC00261 marker for hepatocytes identified here is considered a aetastasis-related lncRNA prognostic signature for colorectal cancer24. To validate the accuracy of the identified marker genes, we constructed two-marker gene panels (Table S3). The UMAP plots clearly illustrate distinct expression patterns of these panels across different cell populations, confirming the precision of the marker gene identification (Fig. 4B). Furthermore, based on the identified markers, combined with the use of the lncPair algorithm, cell type-specific lncRNA and mRNA interaction pairs can be created (Fig. 4C). This not only allows for a deep understanding of how these markers interact to drive cell behavior, but also lays the groundwork for further detailed analysis.

Identification of cell type marker genes. (A) The heatmap illustrates the expression of the identified cell type-specific mRNA (left) and lncRNA (right) across different cell types. (B) UMAP plot demonstrates the identification of various cell types using two marker gene panels. (C) A heatmap is utilized to visually compare and contrast the cell-specific lncRNA-mRNA pairs across diverse cell types, with specific notation of the three most prominent lncRNA-mRNA pairs.
Comparative analysis of tumor samples with different prognoses
Moreover, we conducted a comparative analysis on tumor samples with different prognoses. Firstly, in the context of cellular type proportions, noticeable differences exist between different prognostic tumor samples and adjacent normal samples, among which T cells and Kupffer cells are the most significant. Tumor samples with poorer prognoses had a higher proportion of T cells and a reduced proportion of Kupffer cells, while samples with better prognoses demonstrated the opposite trend (Fig. 5A). Secondly, we measured the difference in gene expression in different cell types based on the Bayesian distance of clustering results. Seven cell types exhibited varying degrees of difference, with B/Plasma cells showing the most significant differentiation, meanwhile T cells and Kupffer cells, which showed evident proportion changes, also exhibited about 1.2 times of variation (Fig. 5B). To delve deeper into the differences in specific genes and functions, we conducted a differential gene analysis on each cell type and performed a Gene Ontology (GO) function enrichment analysis (Fig. 5C,D). Fascinatingly, in the differential genes, we found that the ZBTB16 gene was highly expressed in almost all cell types. ZBTB16 gene mainly participates in the cell cycle process and interacts with histone deacetylase, studies have indicated that it behaves as an oncogene and plays a role in stemness and cell proliferation in colorectal cancer, potentially linking it to prognosis25. From a functional perspective, there was enrichment in aspects related to enzymatic activity regulation and binding, such as GTPase, peptidase, etc. By identifying and comparing the differences in tumor samples of different prognoses, the variability in cellular type proportions and functional changes related to differential prognosis can be unveiled.

Differences in cell type proportions and expression functions between tumor samples with different prognoses. (A) Bar graph illustrates the proportion of cell types in different sample groups. (B) Bhattacharyya distance demonstrates the differences in UMAP clustering among tumor samples with different prognoses. (C) Volcano plot illustrates the upregulated and downregulated genes in different cell types under the comparison of tumor samples with different prognoses, with the top five significantly different genes marked. (D) Dot plot displays the results of the GO functional enrichment of the differentially expressed genes across varying cell types.
Code availability
All software and scripts utilized in this research are publicly accessible, with detailed versions and parameters specified in the Methods section. Where specific parameters are not mentioned, default settings provided by the software developers were applied. The custom scripts used for generating the figures and analyzing the datasets have been uploaded to a GitHub repository, accessible via the following link: https://github.com/chenhongyubio/LongPreservedFFPE.
References
Valderrama-Treviño, A. I., Barrera-Mera, B., Ceballos-Villalva, J. C. & Montalvo-Javé, E. E. Hepatic Metastasis from Colorectal Cancer. Euroasian J. Hepatogastroenterol. 7, 166–175 (2017).
Zeineddine, F. A. et al. Survival improvement for patients with metastatic colorectal cancer over twenty years. NPJ Precis Oncol. 7, 16 (2023).
Martin, J. et al. Colorectal liver metastases: Current management and future perspectives. World J. Clin. Oncol. 11, 761–808 (2020).
El-Sayes, N., Vito, A. & Mossman, K. Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy. Cancers. 13, 806 (2021).
Creasy, J. M. et al. Actual 10-year survival after hepatic resection of colorectal liver metastases: what factors preclude cure? Surgery. 163, 1238–1244 (2018).
Engstrand, J., Nilsson, H., Strömberg, C., Jonas, E. & Freedman, J. Colorectal cancer liver metastases - a population-based study on incidence, management and survival. BMC Cancer. 18, 78 (2018).
Su, Y. M. et al. Five-year survival post hepatectomy for colorectal liver metastases in a real-world Chinese cohort: Recurrence patterns and prediction for potential cure. Cancer Med. 12, 9559–9569 (2023).
Baysoy, A., Bai, Z., Satija, R. & Fan, R. The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 24, 695–713 (2023).
Riegman, P. H. Tissue Preservation and Factors Affecting Tissue Quality. Biobanking of Human Biospecimens: Lessons from 25 Years of Biobanking Experience. 65–80 (2021).
Freitas-Ribeiro, S., Reis, R. L. & Pirraco, R. P. Long-term and short-term preservation strategies for tissue engineering and regenerative medicine products: state of the art and emerging trends. PNAS Nexus. 1, 212 (2022).
Yi, Q. Q. et al. Effect of preservation time of formalin-fixed paraffin-embedded tissues on extractable DNA and RNA quantity. J. Int. Med. Res. 48, 300060520931259 (2020).
Xu, Z. et al. High-throughput single nucleus total RNA sequencing of formalin-fixed paraffin-embedded tissues by snRandom-seq. Nat Commun. 14, 2734 (2023).
Lin, Y. et al. Optimization of FFPE preparation and identification of gene attributes associated with RNA degradation. NAR Genom. Bioinform. 6, lqae008 (2024).
Franzén, O., Gan, L. M. & Björkegren, J. L. M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database. 2019, baz046 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell. 184, 3573–3587 (2021).
Liu, J., Gao, C., Sodicoff, J., Macosko, E. Z. & Welch, J. D. Jointly defining cell types from multiple single-cell datasets using LIGER. Nat. Protoc. 15, 3632–3662 (2020).
Delaney, C. et al. Combinatorial prediction of marker panels from single-cell transcriptomic data. Mol. Syst. Biol. 15, e9005 (2019).
Fan, T. et al. Landscape and functional repertoires of long noncoding RNAs in the pan-cancer tumor microenvironment using single-nucleus total RNA sequencing. bioRxiv, 2023.
Gao, R. et al. Delineating copy number and clonal substructure in human tumors from single-cell transcriptomes. Nat Biotechnol. 39, 599–608 (2021).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb). 2, 100141 (2021).
Single-nucleus transcriptome sequencing of long-preserved FFPE samples from colorectal cancer liver metastasis lesions with different prognoses. National Genomics Data Center Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa-human/browse/HRA007565 (2024).
Single-nucleus transcriptome sequencing of long-preserved FFPE samples from colorectal cancer liver metastasis lesions with different prognoses. figshare https://doi.org/10.6084/m9.figshare.25928743 (2024).
Tang, Q. et al. Discovery and Validation of a Novel Metastasis-Related lncRNA Prognostic Signature for Colorectal Cancer. Front Genet. 13, 704988 (2022).
Iyer, A. S., Shaik, M. R., Raufman, J. P. & Xie, G. The Roles of Zinc Finger Proteins in Colorectal Cancer. Int J Mol Sci. 24, 10249 (2023).
Acknowledgements
We are indebted to all patients who participated in this study. And we extend our heartfelt thanks to Professor Yongcheng Wang from the Liangzhu Experimental Center, Zhejiang University, for his generous support with sequencing technology.
Author information
Authors and Affiliations
Contributions
All the authors participated in the conception and design of the study. H.Y. Chen, X. Zhang, W.B. Han, Q. Cheng, and X.E. Shen obtained and analyzed the data. X. Zhang and W.Q. Jiang collected the samples. H.Y. Chen, and X. Zhang organized the data and drafted the manuscript. L.H. Zeng, L.J. Fan and W.Q. Jiang revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, H., Zhang, X., Cheng, Q. et al. snRNA-seq of long-preserved FFPE samples from colorectal liver metastasis lesions with diverse prognoses. Sci Data 11, 1434 (2024). https://doi.org/10.1038/s41597-024-04323-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-04323-8
