
Abstract
Camellia meiocarpa Hu, a member of Youcha species in the genus Camellia, is an important woody edible Youcha plant with high ecological and economic value. The haplotype-resolved genome assembly of this tetraploid species can shed light on genomic evolution and the functional divergence among subgenomes and haplotypes. In this study, we achieved the first chromosome-level haplotype-resolved genome assembly using PacBio HiFi, Hi-C, and Illumina sequencing. The scaffolds, with an N50 of 44.46 Mb and 41.40 Mb, were mapped to 60 chromosomes and four distinct haplotypes, each with unique transposon features. The haplotypes varied in length (2967.25 Mb to 3041.66 Mb) and contained 51,336 to 52,631 protein-coding genes, 99.4% of which were annotated. Non-coding RNAs and repetitive elements were identified across haplotypes. This comprehensive genomic resource will enhance molecular and genetic studies, aiding in the conservation and utilization of Youcha.
Similar content being viewed by others


Background & Summary
Youcha, in a broad sense, encompasses over more than 60 shrubs belonging to the genus Camellia (Theaceae)1. As one of the world’s most productive woody plants for edible oil, Youcha trees have a cultivation history exceeding 2300 years and possess a diverse wide of uses2,3,4. Among the Camellia species, the Camellia oleifera is the most extensively cultivated and is predominant in oil production, which is valued for its high nutritional content and health benefits5,6,7. Additionally, five other species are cultivated for edible oil production, including C. meiocarpa, C. reticulata, C. chekangoleosa, C. yuhsiensis, and C. vietnamensis3,8. The Youcha trees currently cover more than 4.6 million hectares in South China9. As evergreen and productive oil crops, oil-Camellia trees have garnered increasing attention, especially amidst global food crises.
C. meiocarpa, among the commonly cultivated Youcha plants, is distinguished by its leafy tree habit, large flowers, and abundant fruit (Fig. 1a–d), indicating its potential for agricultural utilization with high oil content and yield10,11,12. It has been reported that the tetraploid C. meiocarpa may have originated from hybridization between closely related diploid species, resulting in an allotetraploid genome13. However, there remains a dearth of chromosomal-scale genomic data supplementation. Polyploidy, a frequent and recurrent phenomenon, is not random and always associated with adaptation to periods of environmental upheaval, providing rich evolutionary material for unique phenotypes14,15,16. In crop improvement, induced polyploidy is considered a powerful tool to increase production, enhance quality, and improve stress adaptation17,18,19. Therefore, it is imperative to explore the genomic feature of Youcha species, particularly those that are naturally polyploid. Within the group of Youcha species, polyploid complexes is frequently observed, from diploid to dodecaploid20,21, yet few high-quality genomes of Youcha species have been published4,6,22,23,24, and none provide a haplotype-scale scenario for genomic divergence.

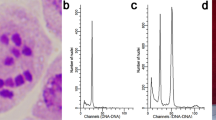
Morphological and genomic characteristics of Camellia meiocarpa Hu. (a) The complete plant and its natural habitat. (b) Close-up of the flower, highlighting the stamen and pistil. (c) A bud in development. (d) The fruit, which is rich in oil. (e) Estimations of genome size and heterozygosity rate based on K-mer counting.
In this study, we used a century-old C. meiocarpa ancient tree, which was identified as a tetraploid after karyotype analysis and ploidy detection and planted at the National Engineering Research Center of Youcha, as the research subject. We constructed and annotated a high-quality chromosome-level reference genome of C. meiocarpa, resolving the four sets of haplotypes for the tetraploid genome (Fig. 2a). The assembly determined a high continuity genome with contig N50s of 44.46 Mb and 41.40 Mb. After anchoring the contigs to 4 × 15 pseudo-chromosomes, the final assembled genome of the four haplotypes were obtained, with lengths of 2967.25 Mb for haplotype A, 3041.66 Mb for haplotype B, 2946.45 Mb for haplotype C, and 3030.54 Mb for haplotype D, covering more than 95% of the K-mer-based estimation of genome size (Table 1; Fig. 1e). A total of 51336, 52010, 51586, and 52631 protein-coding genes were predicted for each of the four haplotypes, of which 99.4% were functionally annotated (Tables 1, 3). The more than 82% of the C. meiocarpa genome is annotated as repetitive sequences (Table 2). This haplotype-resolved tetraploid genome of C. meiocarpa provides an in-depth understanding on the influence of polyploidy on important phenotypic traits and the potential for future utilization in genetic study and breeding programs.

Haplotype assembly and genomic characteristics of Camellia meiocarpa genome. (a) Hi-C interaction heatmap illustrating the scaffolding of the four haplotypes. (b) A circus plot representation of the 4 × 15 chromosomes, detailing their genomic attributes: (I) GC content, (II) transposable element abundance, (III) gene density, (IV) and collinearity blocks among chromosomes. This circos figure was generated using the R package “circlize”75.
Methods
Plant materials and sequencing
C. meiocarpa was cultivated at the National Germplasm Resources Gardern of Youcha (NGRGY) in the Experimental Forestry Farm of Hunan Academy of Forestry (113°01′ E, 28°06′ N). Healthy young leaf samples of C. meiocarpa were collected and subsequently stored in liquid nitrogen. Genomic DNA was extracted from leaf sample by using CTAB method25. Genomic DNA was processed into a Nextera DNA Flex Library, targeting insert sizes between 200 to 400 bp, and assessed for quality and quantity using a Qubit 3.0 Fluorometer and Agilent 2100 system. The library was sequenced on the Illumina Nova-Seq 6000 platform (Illumina, San Diego, CA, USA) to produce paired-end 250 bp reads. The PacBio HiFi library was prepared with the SMRTbell Express Template Prep Kit and sequenced on the PacBio Sequel II platform (Pacifc Biosciences, California, USA), yielding 374.15 Gb HiFi reads with an N50 length of 17.57 kb after removing adapters, assessment of contaminated reads and quality control. For chromosomal scaffolding, young leaf tissues were cross-linked, digested with Dpn II, and ligated. DNA fragments of 300 to 700 bp were selected, and the library was sequenced on the Illumina Nova-Seq platform (Illumina, San Diego, CA, USA) to acquire a total of 383,901,279 paired reads for further chromosomal scaffolding. Transcriptome sequencing was conducted on six different tissues, including young leaf, adult leaf, leaf shoot, stem, bark, and young fruit. The total RNA from these tissues was extracted and sequenced using the Illumina Nova-Seq platform (Illumina, San Diego, CA, USA). Additionally, a merged RNA sample from all six tissues was sequenced on the PacBio Sequel II platform (Pacifc Biosciences, California, USA) for gene structure annotation.
Genome assembly and haplotype-resolved chromosome scaffolding
Initially, we estimated the genome size and heterozygosity rate of C. meiocarpa from HiFi data based on k-mer analysis. We utilized KMC (v3.2.4)26 to count the k-mer frequency at k = 23, and then submitted the results to GenomeScope2 (v2.0.1)27 with the parameter “-k = 23”. Consequently, we determined the genome size of C. meiocarpa to be approximately 3.092 Gb, with a heterozygosity rate of 2.28%.
To resolve the four haplotype sets of the tetraploid C. meiocarpa genome, we employed Hi-C integrated assembly using Hifiasm (v0.18.6) with the default parameters28. The pipeline of purge_dups (v1.2.5) was utilized to remove the redundant haplotigs29, leading to the draft contig-level assemblies with N50 values of 44.46 Mb and 41.40 Mb. After primary filtration using Fastp (v0.23.2)30, the clean Hi-C reads were aligned to the contig-level genome using BWA aligner (v0.7.18) with default parameters31. Only uniquely mapped read pairs were retained for subsequently analysis. The PCR-derived duplicates would be discarded.
Then, the ALLHiC (v0.9.8) workflow32 was applied to classify the contigs and construct the pseudo-chromosome structures. These scaffolding step was conducted to all the contigs from different haplotypes, in order to assign the contigs to correct haplotypes. The pipeline of 3D-DNA (v180419)33 and juicer (v1.6)34 was used to order and orient contigs. Juicerbox (v1.11.08)34 was employed for manual correction of assembly errors, resolving 15 pseudo-chromosomes for each of the four haplotypes (Fig. 2a; Table 1). Finally, we acquired the chromosome-level assemblies of the four haplotypes with final lengths of 2967.25 Mb for haplotype A, 3041.66 Mb for haplotype B, 2946.45 Mb for haplotype C, and 3030.54 Mb for haplotype D (Fig. 2b; Table 1).
Repetitive elements annotation
Repetitive elements were annotated using a combination of de novo and homology-based approaches. First, the LTR_FINDER_parallel35 was ran with the parameters “-threads 16 -harvest_out -size 1000000 -time 300”, while the LTRharvest (v1.62)36 used the parameters “-minlenltr 100 -maxlenltr 7000 -mintsd 4 -maxtsd 6 -motif TGCA -motifmis 1 -similar 85 -vic 10 -seed 20 -seqids yes”. The raw candidates of long terminal repeat retrotransposons (LTR-RTs) identified by LTR_FINDER_parallel and LTRharvest were refined by the LTR_retriever pipeline37. In addition, RepeatModeler (v2.0.4)38 was also used to identify and model de novo transposable element (TE) families. We employed TEcalss (v2.1.3)39 to classify the members from the library merged from the results of LTR_retriever and RepeatModeler. This de novo library was combined with the public repetitive sequence database RepBase (v20181026)40, which was analyzed with RepeatMasker (v4.1.5) and RepeatPriteinMask (v4.1.5)41 for genome-wide identifications of repetitive elements in out haplotype-resolved C. meiocarpa genome.
The analysis revealed that over 2.4 Gb of repetitive sequences were identified in both of the four haplotype sets, constituting more than 82% of the tetraploid C. meiocarpa genome (Table 2). Long terminal repeat retrotransposons (LTR-RTs) predominated among these repetitive elements, comprising 70.57%, 70.81%, 69.31%, and 70.25% of the genomes for haplotypes A, B, C, and D, respectively (Table 2). Furthermore, DNA transposons were found to constitute approximately 10% of the C. meiocarpa genome (Table 2). The substantial proportion of repetitive elements, particularly the proliferation of LTR-RTs, may underlie the ‘genome obesity’ observed in C. meiocarpa.
Gene prediction and function assignment
Gene structures were predicted using an integrated approach of transcriptome-based, ab initio, and homology-based strategies. The repetitive sequences would be masked in the haplotype-resolved genome for gene prediction. In this study, we conducted sequencing of the NGS and full-length transcriptome across six diverse tissues to inform gene prediction (Table S1). The clean NGS reads were aligned to the reference genome of four haplotypes, which encompasses four haplotypes, using Hisat2 (2.2.1) with default parameters42. Stringtie2 (v2.2.1) was then applied to assemble the NGS RNA-seq transcripts43. Furthermore, the PacBio full-length RNA-seq data underwent correction, clustering, and filtering through the IsoSeq3 program within the SMRTlink framework (Pacific Biosciences) (https://github.com/PacificBiosciences/IsoSeq). The Minimap2 aligner44 in conjunction with the cDNA_Cupcake annotator (https://github.com/Magdoll/cDNA_Cupcake) was instrumental in identifying transcripts for subsequent prediction of protein-coding genes. The transcripts from both NGS and full-length RNA-seq were consolidated using the TAMA program (v1.0)45, facilitating the prediction of open reading frames (ORFs) with TransDecoder (v5.7.0)46. The transcripts that encompassed complete ORFs were subsequently designated as transcriptome-based candidates.
For the homology-based gene prediction, we sourced protein-coding genes from five closely related species— C. oleifera6, and C. sinensis47, C. chekiangoleosa22, C. lanceoleosa23, and Arabidopsis thaliana48—to identify homologous regions within the C. meiocarpa genome using tBLASTn49. Subsequently, the Exonerate (v2.4.0) tool50 was utilized to dissect the homology findings, producing a set of homology-based gene candidates.
The gene structures of the C. meiocarpa genome were refined using Augustus (v3.5.0)51 to generate de novo gene annotations. Subsequently, the MAKER (v3.01.03) pipeline52 was employed to synthesize the annotation data from the three distinct sources, complemented by the exclusion of transposon proteins via TransposonPSI (v1.0.0) (https://transposonpsi.sourceforge.net/). Furthermore, we employed the IGV-GSAman program53 to manually refine the gene structures and GFF3toolkit54 to confirm the format of final gff3 annotation file for the four haplotypes of C. meiocarpa. For example, tandem duplicated genes with incomplete exon structures would be discarded (Fig. 3a). Additionally, genes that are specific to a single haplotype and located between pairs of syntenic genes would also be removed based on comparisons among the four haplotypes (Fig. 3b). After the manual corrections of 6,119 genes, the genomes of haplotypes A, B, C, and D were annotated with 51,336, 52,010, 51,586, and 52,631 protein-coding genes, respectively (Fig. 2b; Table 1). Analysis using OMArk and BUSCO55,56 revealed that our gene structure annotations achieved a completeness of over 96% for all four haplotypes (Table S2).

Examples of manual gene structure corrections using IGV-GSAman. (a) Incomplete tandem duplicated genes. Genes with red names are identified as tandem duplicates with incomplete exon structures relative to their upstream and/or downstream genes. These genes would be removed. (b) Simple genes without any syntenic counterparts among the four haplotypes.
For functional annotation of the protein-coding genes, we aligned the protein sequences against various databases using DIAMOND (v2.1.7)57, such as UniProt and the Non-Redundant database, resulting in GO and KOG category annotations (Table 3). We also applied InterProScan (v5.55–88.0)58 and HMMER (v3.3.2)59 to identify motifs and domains, and compared these with the KOfam and Pfam databases60,61, culminating in the acquisition of KEGG pathway annotations (Table 3). Collectively, over 94% of the protein-coding genes across all haplotypes received annotations from at least one functional database, signifying the high quality of annotations within the C. meiocarpa genome.
Additionally, non-coding RNA in the C. meiocarpa genome was annotated, encompassing miRNA, tRNA, rRNA, and snRNA (Table 1). The tRNA structure was discerned using tRNAscan-SE (v2.0.12)62, while rRNA was forecasted using RNAmmer (v1.2)63. The remaining non-coding RNAs were categorized through the INFERNAL (v1.1.4)64 and Rfam database (v14.9)65.
Haplotype clustering
Recently, the haplotype-resolved genome of tetraploid C. oleifera has been reported66, providing valuable insights into the evolution, agronomic trait development, and genetic architecture of oil Camellia plants. The genomic relationship between the tetraploid C. meiocarpa and C. oleifera could be explored based on the analyses of genomic content. Based on the LTR_retriever outcomes, we conducted clustering on the complete set of 120 chromosomes to confirm the partitions of the haplotypes of both C. meiocarpa and C. oleifera (Fig. 4). Utilizing the SSM method67, we constructed a similarity matrix for these chromosomes. Our findings reveal that the four haplotypes of C. oleifera form a distinct cluster, yet they exhibit genomic contents that are notably different from those of C. meiocarpa (Fig. 4). These results provide robust evidence supporting the classification of C. meiocarpa and C. oleifera as two separate species at the genomic level. The chromosomes of C. meiocarpa can be distinctly categorized into four haplotype groups, which implies their independent evolutionary origins. This genetic partitioning implies that the tetraploid genome of C. meiocarpa may have arisen from two distinct hybridization events, which have resulted in varied LTR similarities among the homologous chromosome sets.

Cluster analysis of the chromosomes based on LTR similarity. The tetraploid geonomes of Camellia oleifera and C. meiocarpa, are investigated here, suggesting the relationships and genomic structures between these two species.
Data Records
The PacBio HiFi long reads, Hi-C interaction data, and multi-tissue RNA-seq datasets RNA-seq data of multiple tissues have been submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database under the SRA accession number SRP53126768. The final chromosome assembly has been deposited at ENA under the accession number GCA_965213565.169, as well as at National Genomics Data Center (accession GWHFIBF0000000070). The genome assembly and annotation results have also been deposited in the figshare71.
Technical Validation
The continuity of the genome assembly was evaluated by remapping NGS and HiFi genomic reads to the assembled haplotype-resolved genome (Fig. 5a). The NGS reads were aligned using BWA (v0.7.17) program31, achieving high coverage rates of over 99.9% for all haplotypes. For the HiFi reads, Minimap2 aligner44 was utilized to assess coverage at minimum depths of 1x, 10x, and 20x. The results demonstrated that more than 99.9% of the assembled genome was covered at least 1x for all four haplotypes, with approximately 95% and 83% of the genome covered at 10x and 20x depths, respectively (Fig. 5a).

Evaluations of genome assembly quality. (a) Mapping rates of NGS and HiFi reads to the assembled genomes, demonstrating the coverage of the assembled haplotype genomes. (b) BUSCO analysis results for each haplotype, indicating the completeness of assembly.
The completeness of the genome assembly was assessed using BUSCO (v5.2.2) with the embryophyta_odb10 orthologous database, which contains 1,614 conserved single-copy genes56. The BUSCO analysis indicated a completeness range of 94 to 95.2% at the chromosome level for the four haplotypes (Fig. 5b). Additionally, the LTR Assembly Index (LAI) was calculated using LTR_retriever37,72, yielding values between 12.13 and 15.49. These assessments confirm that the haplotype-resolved genome of C. meiocarpa exhibits excellent assembly quality, characterized by high completeness and continuity.
We further assessed genome completeness using Merqury (v1.3) program73 with HiFi reads, achieving a high-quality value (QV) of 75.90. Additionally, we utilized CRAQ (Clipping information for Revealing Assembly Quality)74 to generate further evaluations of the genome assembly quality. These analyses indicated an R-AQI (Reference Assembly Quality Index) of 94.67 and an S-AQI (Scaffold Assembly Quality Index) of 92.97, both of which are indicative of a high-quality genome assembly.
Code availability
All software and pipelines were executed following the manuals and protocols of the respective published bioinformatics tools. The versions and parameters of the software are described in the Methods section. No custom code was used in this study.
References
Chen, J., Guo, Y., Hu, X. & Zhou, K. Comparison of the Chloroplast Genome Sequences of 13 Oil-Tea Camellia Samples and Identification of an Undetermined Oil-Tea Camellia Species From Hainan Province. Frontiers in Plant Science 12 (2022).
Yan, H. et al. Assessment of the Genetic Relationship and Population Structure in Oil-Tea Camellia Species Using Simple Sequence Repeat (SSR) Markers. Genes 13 (2022).
Yu, J., Yan, H., Wu, Y., Wang, Y. & Xia, P. Quality Evaluation of the Oil of Camellia spp. Foods 11 (2022).
Zhu, H. et al. The complex hexaploid oil-Camellia genome traces back its phylogenomic history and multi-omics analysis of Camellia oil biosynthesis. Plant Biotechnology Journal n/a, https://doi.org/10.1111/pbi.14412 (2024).
Gao, L. et al. Recent advances in the extraction, composition analysis and bioactivity of Camellia (Camellia oleifera Abel.) oil. Trends in Food Science & Technology 143, 104211, https://doi.org/10.1016/j.tifs.2023.104211 (2024).
Lin, P. et al. The genome of oil-Camellia and population genomics analysis provide insights into seed oil domestication. Genome Biology 23, 14, https://doi.org/10.1186/s13059-021-02599-2 (2022).
Ma, J., Ye, H., Rui, Y., Chen, G. & Zhang, N. Fatty acid composition of Camellia oleifera oil. Journal für Verbraucherschutz und Lebensmittelsicherheit 6, 9–12, https://doi.org/10.1007/s00003-010-0581-3 (2011).
Yang, C., Liu, X., Chen, Z., Lin, Y. & Wang, S. Comparison of Oil Content and Fatty Acid Profile of Ten New Camellia oleifera Cultivars. Journal of Lipids 2016, 3982486, https://doi.org/10.1155/2016/3982486 (2016).
Luan, F. et al. Recent advances in Camellia oleifera Abel: A review of nutritional constituents, biofunctional properties, and potential industrial applications. Journal of Functional Foods 75, 104242, https://doi.org/10.1016/j.jff.2020.104242 (2020).
Feng, J.-L. et al. Transcriptome comparative analysis of two Camellia species reveals lipid metabolism during mature seed natural drying. Trees 31, 1827–1848, https://doi.org/10.1007/s00468-017-1588-5 (2017).
Huang, J. Correlation of fruit characters and oil content and fatty acid composition of Camellia meiocarpa. China Oils and Fats (2013).
Chen, M., Zhang, Y., Du, Z., Kong, X. & Zhu, X. Integrative Metabolic and Transcriptomic Profiling in Camellia oleifera and Camellia meiocarpa Uncover Potential Mechanisms That Govern Triacylglycerol Degradation during Seed Desiccation. Plants 12 (2023).
Qin, S.-Y. et al. Phylogenomic insights into the reticulate evolution of Camellia sect. Paracamellia Sealy (Theaceae). Journal of Systematics and Evolution 62, 38–54, https://doi.org/10.1111/jse.12948 (2024).
Fox, D. T., Soltis, D. E., Soltis, P. S., Ashman, T.-L. & Van de Peer, Y. Polyploidy: A Biological Force From Cells to Ecosystems. Trends in Cell Biology 30, 688–694, https://doi.org/10.1016/j.tcb.2020.06.006 (2020).
Heslop-Harrison, J. S., Schwarzacher, T. & Liu, Q. Polyploidy: its consequences and enabling role in plant diversification and evolution. Annals of Botany 131, 1–10, https://doi.org/10.1093/aob/mcac132 (2023).
Van de Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nature Reviews Genetics 18, 411–424, https://doi.org/10.1038/nrg.2017.26 (2017).
Alam, H., Razaq, M. & Salahuddin Induced Polyploidy as a Tool for Increasing Tea (Camellia sinensis L.) Production. Journal of Northeast Agricultural University (English Edition) 22, 43–47, https://doi.org/10.1016/S1006-8104(16)30005-8 (2015).
Gantait, S. & Mukherjee, E. Induced autopolyploidy—a promising approach for enhanced biosynthesis of plant secondary metabolites: an insight. Journal of Genetic Engineering and Biotechnology 19, 4, https://doi.org/10.1186/s43141-020-00109-8 (2021).
Sabooni, N. & Gharaghani, A. Induced polyploidy deeply influences reproductive life cycles, related phytochemical features, and phytohormonal activities in blackberry species. Frontiers in Plant Science 13 (2022).
Li, Y. et al. Ploidy and fruit trait variation in oil-tea Camellia: Implications for ploidy breeding. Journal of Integrative Agriculture 23, 2662–2673, https://doi.org/10.1016/j.jia.2024.03.016 (2024).
Ye, T., Li, S., Li, Y., Xiao, S. & Yuan, D. Impact of polyploidization on genome evolution and phenotypic diversity in oil-tea Camellia. Industrial Crops and Products 218, 118928, https://doi.org/10.1016/j.indcrop.2024.118928 (2024).
Shen, T.-f et al. The reference genome of Camellia chekiangoleosa provides insights into Camellia evolution and tea oil biosynthesis. Horticulture Research 9, uhab083, https://doi.org/10.1093/hr/uhab083 (2022).
Gong, W. et al. Chromosome-level genome of Camellia lanceoleosa provides a valuable resource for understanding genome evolution and self-incompatibility. The Plant Journal 110, 881–898, https://doi.org/10.1111/tpj.15739 (2022).
Zhang, F., Feng, L.-y, Lin, P.-f, Jia, J.-j & Gao, L.-z Chromosome-scale genome assembly of oil-tea tree Camellia crapnelliana. Scientific Data 11, 599, https://doi.org/10.1038/s41597-024-03459-x (2024).
Allen, G. C., Flores-Vergara, M. A., Krasynanski, S., Kumar, S. & Thompson, W. F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nature Protocols 1, 2320–2325, https://doi.org/10.1038/nprot.2006.384 (2006).
Kokot, M., Długosz, M. & Deorowicz, S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761, https://doi.org/10.1093/bioinformatics/btx304 (2017).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432, https://doi.org/10.1038/s41467-020-14998-3 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898, https://doi.org/10.1093/bioinformatics/btaa025 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nature Plants 5, 833–845, https://doi.org/10.1038/s41477-019-0487-8 (2019).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Ou, S. & Jiang, N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mobile DNA 10, 48, https://doi.org/10.1186/s13100-019-0193-0 (2019).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiology 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330, https://doi.org/10.1093/bioinformatics/btp084 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11, https://doi.org/10.1186/s13100-015-0041-9 (2015).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current Protocols in Bioinformatics 25, 4.10.11–14.10.14, https://doi.org/10.1002/0471250953.bi0410s25 (2009).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biology 20, 278, https://doi.org/10.1186/s13059-019-1910-1 (2019).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100, https://doi.org/10.1093/bioinformatics/bty191 (2018).
Kuo, R. I. et al. Illuminating the dark side of the human transcriptome with long read transcript sequencing. BMC Genomics 21, 751, https://doi.org/10.1186/s12864-020-07123-7 (2020).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512, https://doi.org/10.1038/nprot.2013.084 (2013).
Chen, J.-D. et al. The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant. Horticulture Research 7, 63, https://doi.org/10.1038/s41438-020-0288-2 (2020).
Cheng, C.-Y. et al. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. The Plant Journal 89, 789–804, https://doi.org/10.1111/tpj.13415 (2017).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421, https://doi.org/10.1186/1471-2105-10-421 (2009).
Slater, G. S. C. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31, https://doi.org/10.1186/1471-2105-6-31 (2005).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–W439, https://doi.org/10.1093/nar/gkl200 (2006).
Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome Annotation and Curation Using MAKER and MAKER-P. Current Protocols in Bioinformatics 48, 4.11.11–14.11.39, https://doi.org/10.1002/0471250953.bi0411s48 (2014).
Thorvaldsdóttir, H., Robinson, J. T. & Mesirov, J. P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics 14, 178–192, https://doi.org/10.1093/bib/bbs017 (2013).
Chen, M.-J. M., Lin, H., Chiang, L.-M., Childers, C. P. & Poelchau, M. F. in Insect Genomics: Methods and Protocols (eds Susan J. Brown & Michael E. Pfrender) 75–87 (Springer New York, 2019).
Nevers, Y. et al. Quality assessment of gene repertoire annotations with OMArk. Nature Biotechnology https://doi.org/10.1038/s41587-024-02147-w (2024).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212, https://doi.org/10.1093/bioinformatics/btv351 (2015).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240, https://doi.org/10.1093/bioinformatics/btu031 (2014).
Potter, S. C. et al. HMMER web server: 2018 update. Nucleic Acids Research 46, W200–W204, https://doi.org/10.1093/nar/gky448 (2018).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252, https://doi.org/10.1093/bioinformatics/btz859 (2020).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Research 49, D412–D419, https://doi.org/10.1093/nar/gkaa913 (2021).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Research 49, 9077–9096, https://doi.org/10.1093/nar/gkab688 (2021).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Research 49, D192–D200, https://doi.org/10.1093/nar/gkaa1047 (2021).
Zhang, L. et al. The tetraploid <em>Camellia oleifera</em> genome provides insights into evolution, agronomic traits, and genetic architecture of oil <em>Camellia</em> plants. Cell Reports 43, https://doi.org/10.1016/j.celrep.2024.114902 (2024).
Lyu, H., Ou, S., Yim, W. C. & Yu, Q. Deciphering octoploid strawberry evolution with serial LTR similarity matrices for subgenome partition. bioRxiv, 2024.2007.2031.606053, https://doi.org/10.1101/2024.07.31.606053 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP531267 (2024).
ENA https://identifiers.org/ncbi/insdc.gca:GCA_965213565.1 (2025).
NGDC/CNCB https://ngdc.cncb.ac.cn/gwh/Assembly/86772/show (2024).
Wang, R. et al. Haplotype-resolved genome assembly of the tetraploid Youcha tree Camellia meiocarpa Hu, figshare. Dataset https://doi.org/10.6084/m9.figshare.26926918.v2 (2024).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Research 46, e126–e126, https://doi.org/10.1093/nar/gky730 (2018).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Li, K., Xu, P., Wang, J., Yi, X. & Jiao, Y. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nature Communications 14, 6556, https://doi.org/10.1038/s41467-023-42336-w (2023).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812, https://doi.org/10.1093/bioinformatics/btu393 (2014).
Acknowledgements
This work was supported by the Top Ten Technological Research Projects in Hunan Province (2024NK1020), R&D Plan for Key Areas in Hunan Province (2023NK2005) and Major Special Project of Changsha Science and Technology Bureau (KQ2102007).
Author information
Authors and Affiliations
Contributions
Yongzhong Chen conceived and designed the study and revised the manuscript. Haomin Lyu and Zhilong He write the manuscript. Weiguo Li, Chengfeng Xun, Yushen Ma and Gaohong Xiao analyze the data. Rui Wang and Changrong Ye revise the manuscript. Xiangnan Wang, Ying Zhang and Zhen Zhang collected samples and performed experiments. Bingchuan Tian and Gaofeng Jia supervise this study. All authors read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, R., Li, W., He, Z. et al. Haplotype-resolved genome assembly of the tetraploid Youcha tree Camellia meiocarpa Hu. Sci Data 12, 541 (2025). https://doi.org/10.1038/s41597-025-04887-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04887-z
