
Abstract
Invasive species pose a serious threat to ecosystems and biodiversity, leading to considerable economic losses for countries. The papaya mealybug (Paracoccus marginatus), is a prominent invasive pest that affects over 200 plant species and has been recorded in more than 60 countries and regions.Here, the chromosome-level genome of P. marginatus was assembled using PacBio and Hi-C technologies. The resulting genome, with a total size of 213.81 Mb, was organized into four chromosomes. The contig and scaffold N50 values were 20.2 Mb and 48.01 Mb, respectively. The genome assembly attained a BUSCO completeness score of 95.5%, and CEGMA analysis showed that 99.56% of the genome was thoroughly annotated. It includes 13,367 predicted protein-coding genes, with 49.26% of the assembly identified as repetitive sequences. This high-quality genome serves as a valuable resource for a range of research fields, such as population genetics, evolutionary studies, invasive species management, and comparative genomics within Hemiptera and other insect groups.
Similar content being viewed by others


Background & Summary
The papaya mealybug, Paracoccus marginatus Williams and Granara de Willink (Hemiptera: Pseudococcidae) (Fig. 1), is a highly polyphagous pest that affects more than 200 plant species1. It poses significant economic risks to various crops and ornamental plants, such as avocado (Persea americana)2, cassava (Manihot esculenta)3, papaya (Carica papaya)4, tomato (Solanum lycopersicum)5, chili pepper (Capsicum annuum)5, and eggplant (Solanum melongena)6. Originally native to Mexico and Central America7,8, P. marginatus was not a major pest in its native habitat, likely due to the presence of endemic natural enemies. However, it became a serious pest after its introduction to the Caribbean in the 1990s, and has since spread to over 60 countries, including tropical and subtropical regions in the Americas, Africa, and Asia1,7,9. Studies have shown that infestations in papaya orchards in Bangladesh result in average economic losses of approximately USD 700 per hectare annually due to P. marginatus damage10. In Kenya, the papaya mealybug causes losses exceeding USD 29.8 million per year, with individual households experiencing losses between USD 51 and USD 740, depending on local conditions and market access11. Crop losses due to P. marginatus can range from 10% to 60% depending on the crop, but in severe cases, they can exceed 90%12,13,14. These significant economic and yield losses highlight the global threat posed by the papaya mealybug, making it a critical concern for agricultural sustainability and food security.

Ecological photos of papaya mealybug. (A) adult female. (B) adult male; (C) eggs; (D) feeding on papaya. (Collected site: Sanya city, Hainan Province, China, June 2022; Images were provided by Linjia Xue, the one of author).
Recent studies have revealed that P. marginatus in Asia consists of a single haplotype, suggesting that its presence in the region is the result of a recent invasion (unpublished data)8. This could be attributed to the short length of the gene segments used, which might not capture sufficient genetic diversity. Therefore, longer molecular markers are needed for more comprehensive future studies. The lack of high-quality genomic information has limited our understanding of the molecular mechanisms underlying P. marginatus ecology and the development of new molecular markers. While extensive research has been conducted on biological control15,16, risk assessment7, biology17, and genetics8, the molecular mechanisms behind the papaya mealybug’s adaptability to various environments and host plants remain unclear.
In the current study, a chromosome-scale genome assembly for papaya mealybug was achieved by integrating Hi-Fi sequencing technologies with the Hi-C scaffolding approach. The assembled genome had contig and scaffold N50s of 20.2 Mb and 48.01 Mb, respectively. The assembled genome, with a total size of 213.81 Mb, was anchored onto four chromosomes. The genome exhibited contig and scaffold N50s of 20.2 Mb and 48.01 Mb, respectively. A total of 13,367 protein-coding genes were predicted, with 11,719 of these genes functionally annotated.
Methods
Sampling and sequencing
The papaya mealybugs used for sequencing were collected from Sanya City, Hainan Province, China, in June 2022. These mealybugs were not exposed to any insecticides and were found on papaya (Carica papaya L.). The specimens were reared in the laboratory at the College of Plant Protection, Shanxi Agricultural University. A total of 150 female adult individuals were used for genome sequencing, with the entire bodies of these samples utilized to construct the library.
High-quality DNA was extracted from the whole body of adult female by the QIAGEN DNeasy Blood and Tissue kit (Qiagen, Germantown, USA). Illumina and third generation sequencing (PacBio) were used respectively. An excellent integrity of DNA molecules was observed in the current study. Genome sequencing was carried out by Biomaker Biotechnology Company in Beijing. The raw data will be used in the laboratory for subsequent genome assembly, annotation, and other related tasks.
Genome size estimation and contig assembly
A genome survey of the papaya mealybug was performed using K-mer analysis (K = 19) with a 350 bp library dataset. The Illumina short-read DNA library produced 41.31 Gb of high-quality filtered data, providing 143.69 × genome coverage, with Q20 and Q30 scores exceeding 99.21% and 95.43%, respectively. Based on the 19-mer frequency distribution analysis (Fig. 2), the papaya mealybug genome was estimated to be approximately 287.46 Mb (287,457,296 bp) in length. The heterozygosity of the genome was calculated to be 0.57%. Repetitive sequences constituted 49.26% of the genome, and the GC content was 32.82%.

The distribution of 19-mer frequency in the papaya mealybug. Frequency distribution of k-mers of different occurrences in two paired-end libraries. K-mer occurrences (x-axis) were plotted against their frequencies (y-axis).
De novo assembly of the PacBio HiFi reads was performed using Hifiasm (version 0.16)18, with redundant sequences filtered out using purge_dups19. The completeness of the genome was assessed using CEGMA (version 2.5)20 and BUSCO (version 4)21. A total of 14.01 Gb of HiFi reads, with a maximum read length of 43,103 bp and an average read length of 14,691 bp, were obtained (Tables S1, S2). The initial genomic contig sequence had a total length of 245.75 Mb and a contig N50 of 20.02 Mb. After removing redundancy, the length of the genomic contig was reduced to 215.04 Mb, maintaining a contig N50 of 20.02 Mb (Table S2). CEGMA assessment indicated a completeness of 99.56%, while BUSCO analysis yielded a completeness score of 95.50% based on the Hemiptera database in OrthoDB 10.
Hi-C analysis and chromosome assembly
Hi-C libraries were constructed and sequenced using the Illumina NovaSeq. 6000 platform (Biomaker Technologies Co., Ltd., Beijing, China). Burrows-Wheeler Aligner (version 0.7.17-r1188) was used to align the paired-end clean reads to the assembled genome sequence, yielding uniquely mapped read pairs. Final valid reads for downstream analysis were selected after removing invalid read pairs, including dangling ends, self-cycles, re-ligation, and discarded products, using Hic-Pro (version 2.10.0). The chromosome matrix was visualized as a heatmap, displaying diagonal patches indicative of strong linkages. Gene density, transposable element (TE) density, and GC content were plotted as a circus plot using the Circlize package (version 0.4.1)22.
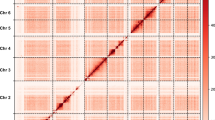
A total of 208,871,684 read pairs (61.87 Gb of clean data; 42 × genome coverage) were generated from the Hi-C library (Table S1). Of these, 82.96% (173,267,369 bp) were uniquely mapped to the assembled genome. Among the uniquely mapped read pairs, 64.25% (111,328,377 bp) were valid interaction pairs used for the Hi-C assembly. The Hi-C assembly anchored 213.81 Mb (213,808,492 bp) of the genome sequences to 4 chromosomes, with the order and direction of 213.54 Mb (99.87%) of these sequences accurately determined (Table 1, Fig. 3A). The heatmap of the Hi-C interaction bins confirms the high quality of the genome assembly (Fig. 3B). Detailed chromosome sequence distribution is provided in Table S3. The final assembled genome size for P. marginatus is 213.81 Mb, consisting of 98 contigs and 31 scaffolds, with contig and scaffold N50 values of 20.2 Mb and 48.01 Mb, respectively (Table 1).

Heatmap of genome-wide Hi-C data and overview of the genomic landscape of P. marginatus. (A) Circos plot of distribution of the genomic elements in papaya mealybug was visualized by Circos. Blocks on the outmost circle represent all four chromosomes of P. marginatus. Peak plots from outer to inner circles represent the length of each chromosome, TE density, Gene density and GC content. (B) The heatmap showing all-by-all Hi-C interactions among four pseudochromosomes of P. marginatus. The frequency of Hi-C interaction links is represented by colours, which ranges from white (low) to red (high).
Genome annotation
The repetitive elements in the papaya mealybug genome were identified and annotated using RepeatModeler2 (version 2.0.1)23, which includes two tools: RECON (version 1.0.8)24 and RepeatScout (version 1.0.6)25. The combined results from RepeatModeler, along with databases Repbase (v19.06)26, REXdb (v3.0)27, and Dfam (version 1.07)28, were used to create the final repeat sequence libraries for subsequent analysis with RepeatMasker (v4.1.2)29. Tandem repeats (TRs) were also identified using MISA (Microsatellite Identification tool) (v2.1) and TRF (Tandem Repeat Finder) (v4.09). In total, 55,387,198 bp of transposable elements (TEs) were annotated, representing 25.76% of the entire genome (Table S4). Among these TEs, DNA transposons comprised 22.79% of the whole genome, making them the largest category of repeats. Tandem repeat sequences accounted for 11,202,955 bp, which is 5.21% of the total TEs (Table S5).
Genome gene structure prediction was carried out using a combination of three approaches: ab initio, homology-based, and RNA-seq-based methods. Ab initio gene models were predicted using Augustus (version 3.1.0)30 and SNAP (version 2006-07-08)31. Homology-based predictions were performed with GeMoMa (version 1.7)32, utilizing homologous genes from three species: Aphis gossypii, Phenacoccus solenopsis, and Metopolophium dirhodum. GeneMarkS-T (version 5.1)33 was employed for predictions based on assembled transcripts, while PASA (version 2.4.1)34 was used to refine gene predictions based on unigenes assembled by Trinity (version 2.11)35. The final gene models were integrated using EVM (version 1.1.1)36 and further modified with PASA (version 2.4.1). The completeness of the genome prediction for the papaya mealybug was evaluated using BUSCO (version 5.2.2)21 by searching against the Hemiptera database. Additionally, various non-coding RNAs were identified, including tRNAs, rRNAs, miRNAs, snRNAs, and snoRNAs. tRNAs were determined using tRNAscan-SE (version 1.3.1)37; rRNAs were predicted with barrnap (version 0.9)38; miRNAs, snoRNAs, and snRNAs were identified using Infernal (version 1.1)39 based on the Rfam (version 14.5)40 database. Pseudogenes were searched using GenBlastA (version 1.0.4)41 and GeneWise (version 2.4.1).
A total of 13,367 protein-coding genes were predicted by integrating ab initio, homology-based, and RNA-seq strategies (Table S6, Fig. 4). The average gene length is 6,164 bp, with an average exon length of 2,001 bp, average coding sequence length of 1,524 bp, and average intron length of 4,162 bp (Table 2, Fig. 4). Additionally, 2,346 BUSCOs were identified, with a completeness of 93.47%, 0.52% fragmentation (13 BUSCOs), and 6.02% missing (151 BUSCOs), indicating a high accuracy in gene prediction across the papaya mealybug genome. Non-coding RNAs were found to have an average length of 246 bp, including 32 rRNA, 195 tRNA, 7 miRNA, and 13 snRNA (Table S7). A total of 10 pseudogenes were identified, with a combined length of 16,406 bp and an average length of 1,640.6 bp (Table S7).

The protein-coding genes were predicted by integrating three methods. Venn diagram illustrate protein-coding genes were predicted based on 3 different strategies.
Gene function prediction was conducted using various databases, including NR, eggNOG (Evolutionary Genealogy of Genes: Non-supervised Orthologous Groups)42, KEGG (Kyoto Encyclopedia of Genes and Genomes)43, TrEMBL, KOG, SWISS-PROT, and Pfam44. Motifs and domains were identified using InterProScan (version 5.34–73.0)45. In total, 11,719 protein-coding genes in the papaya genome were functionally annotated through alignments with these public databases (NR, eggNOG, GO, KEGG, Swiss-Prot, Pfam, KOG, and TrEMBL) (Table S8). Additionally, 591 motifs and 19,572 domains were annotated in this study.
Data Records
The PacBio and Hi-C sequencing data that were used for the genome assembly have been deposited in the NCBI Sequence Read Archive with accession number SRR3255413446 and SRR2549786647 under BioProject accession number PRJNA992385. The final chromosome assembly were deposited in the GenBank with accession number JBLZST00000000048. The annotation results of repeated sequences, gene structure and functional prediction were deposited in the Figshare database49.
Technical Validation
DNA quality was evaluated by 0.75% agarose gel electrophoresis, and DNA concentration was quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher, USA) in combination with a Qubit 3.0 Fluorometer (Invitrogen, USA).
Genome completeness was assessed using CEGMA (v2.5)20 and BUSCO (v4.0)21. The final assembly spanned 213.81 Mb, with 99.56% of sequences anchored to four chromosomes. Assembly continuity metrics revealed contig N50 of 20.2 Mb and scaffold N50 of 48.01 Mb. Benchmarking analyses demonstrated high completeness: BUSCO assessment yielded a score of 95.5%, while CEGMA identified 99.56% core eukaryotic genes, collectively supporting the high-quality annotation of P. marginatus genome.
Code availability
All software and pipelines were executed according to the manual and protocols of the published bioinformatic tools. The version and code/parameters of software have been described in Methods.
References
García Morales, M et al. Hardy. ScaleNet: A literature-based model of scale insect biology and systematics. Database. http://scalenet.info (2016).
Miller, D. R. & Miller, G. L. Redescription of Paracoccus marginatus Williams and Granara de Willink (Hemiptera: Coccoidea: Pseudococcidae), including descriptions of the immature stages and adult male. Proc. Entomol. Soc. Wash. 104(1), 1–23 (2002).
Matile-Ferrero, D., Étienne, J. & Tiego, G. Introduction de deux ravageurs d’importance pour la Guyane française: Maconellicoccus hirsutus et Paracoccus marginatus (Hem., Coccoidea, Pseudococcidae). Bull. Soc. Entomol. Fr. 105(5), 485–486 (2000).
Kansiime, M. K. et al. Crop losses and economic impact associated with papaya mealybug (Paracoccus marginatus) infestation in Kenya. Int. J. Pest Manag. 69(2), 150–163 (2023).
Galanihe, L. D. et al. Occurrence, distribution and control of papaya mealybug, Paracoccus marginatus (Hemiptera: Pseudococcidae), an invasive alien pest in Sri Lanka. Trop. Agric. Res. Ext. 13(3), 81–86 (2011).
Sakthivel, P., Karuppuchamy, P., Kalyanasundaram, M. & Srinivasan, T. Host Plants of Invasive Papaya Mealybug, Paracoccus marginatus (Williams and Granara de Willink) in Tamil Nadu. Madras Agri. J. 99(7-9), 615–619 (2012).
Finch, E. A. et al. The potential distribution global distribution of the papaya mealybug, Paracoccus marginatus, a polyphagous pest. Pest Manag. Sci. 77(3), 1361–1370 (2020).
Ahmed, M. Z. et al. First report of the papaya mealybug, Paracoccus marginatus (Hemiptera: Pseudococcidae), in China and genetic record for its recent invasion in Asia and Africa. Fla Entomol. 98(4), 1157–1162 (2015).
Li, J. Y. et al. Shi. Sublethal and Transgenerational toxicities of chlorfenapyr on biological traits and enzyme activities of Paracoccus marginatus (Hemiptera: Pseudococcidae). Insects 13(874), 1–13 (2022).
Khan, M., Biswas, M., Ahmed, K. & Sheheli, S. Outbreak of Paracoccus marginatus in Bangladesh and its control strategies in the fields. Pro. Agri. 25, 17–22 (2015).
Ndlela, S., Niassy, S. & Mohamed, S. A. Important alien and potential native invasive insect pests of key fruit trees in Sub-Saharan Africa: advances in sustainable pre-and post-harvest management approaches. CABI 3(7), 1–46 (2022).
Myrick, S., Norton, G. W., Selvaraj, K. N., Natarajan, K. & Muniappan, R. Economic impact of classical biological control of papaya mealybug in India. Crop Prot. 56, 82–86 (2014).
Chen, Q. et al. Resistant cassava cultivars inhibit the papaya mealybug Paracoccus marginatus population based on their interaction: from physiological and biochemical perspectives. J. Pest Sci. 96, 555–572 (2023).
Macharia, I. et al. First report and distribution of the papaya mealybug, Paracoccus marginatus, in Kenya. J. Agri. Urban Entomol. 33, 142–150 (2017).
Mani, M., Shylesha, M. & Shivaraju, C. First report of the invasive papaya mealybug, Paracoccus marginatus Williams & Granara de Willink (Homoptera: Pseudococcidae) in Rajasthan. Pest Manag. Hor. Eco. 18(2), 234–234 (2012).
Lem, K. H., Tranm, T. H. D., Tranm, D. H. & Doanm, C. V. Parasitoid wasp Acerophagus papayae: a promising solution for the control of Papaya mealybug Paracoccus marginatus in cassava fields in Vietnam. Insects 14(6), 1–12 (2023).
Chuai, H. Y., Shi, M. Z., Li, J. Y., Zheng, L. Z. & Fu, J. W. Fitness of the Papaya mealybug, Paracoccus marginatus (Hemiptera: Pseudococcidae), after transferring from Solanum tuberosum to Carica papaya, Ipomoea batatas, and Alternanthern philoxeroides. Insects 13(804), 1–16 (2022).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Guan, D. F. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36(9), 2896–2898 (2020).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Gu, Z., Gu, L., Eils, R., Schlesner, M. & Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 30(19), 2811–2812 (2014).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNA 117(17), 9451–9457 (2020).
Bao, Z. R. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12(8), 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Bao, W. D., Kojima, K. K. & Kohany, O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6(11), 1–6 (2015).
Neumann, P., Novak, P., Hostakova, N. & Macas, J. Systematic survey of plant LTR- retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mobile DNA 10(1), 1–17 (2019).
Wheeler, T. J. et al. Dfam: a database of reprtitive DNA based on profile hidden Markov models. Nucleic Acids Res. 41(D1), D70–D82 (2013).
Tarailo-Graovac, M. & Chen, N. S. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in Bioinformatics, chapter 4, 4.10.1–4.10.14 (2009).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntonically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24(5), 637–644 (2008).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5(59), 1–9 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44(9), 1–11 (2016).
Tang, S. Y. Y., Lomsadze, A. & Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 43(12), 1–10 (2015).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31(19), 5654–5666 (2003).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat. Biotech. 29(7), 644–652 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EvidenceModeler and the program to assemble spliced alignments. Genome Biol. 9(1), 1–22 (2008).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25(5), 955–964 (1997).
Torkel, L. A novel method for predicting ribosomal RNA genes in Prokaryotic genomes. Degree projects in Bioinformatics. (2017).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29(22), 2933–2935 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33(1), D121–D124 (2005).
Shen, R., Chu, J. S. C., Wang, K., Pei, J. & Chen, N. S. genBlastA: BLAST to identify homologous gene sequences. Genome Res. 19, 143–149 (2009).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47(D1), D309–D314 (2019).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44(D1), D457–D462 (2015).
Finn, R. D. et al. Pfam: clans, web tools and services. Nucleic Acids Res. 34(D1), D247–D251 (2006).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9), 1236–1240 (2014).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25497866 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR32554134 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBLZST000000000 (2025).
Wei, J.-F. A chromosome-level genome assembly of important alien species Paracoccus marginatus based on PacBio and Hi-C technologies. Figshare. https://doi.org/10.6084/m9.figshare.24078891 (2024).
Acknowledgements
This research was supported by National Natural Science Foundation of China (No. 32100370), Science and Technology Innovation Funds of Shanxi Agricultural University (2020BQ79), the Excellent Doctoral Award of Shanxi Province for Scientific Research Project (SXBYKY2021024), Science and Technology Innovation Projects of Universities in Shanxi Province (2021L097), Natural Science Foundation of Shanxi Province (202103021224132), Key research and development program of Hainan Province (ZDYF2020088), the earmarked fund for Modern Agro-industry Technology Research System(2023CYJSTX07-03 and the Program for the Top Young Talents of Shanxi Province (2018).
Author information
Authors and Affiliations
Contributions
Jiufeng Wei: Validation, Formal analysis, Investigation, Figure development, Writing draft; Bo Cai: Data curation, Resources, Investigation, Writing draft; Linjia Xue: Resources; Gaoxiang Zhang: Writing review, Data curation; Qing Zhao: Data curation; YunYun Lu: Validation; Minmin Niu: Methodology, Project administration, Writing review, funding; Wei Ji: onceptualization, Methodology, Project administration, Funding, editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wei, J., Xue, J., Shen, X. et al. Chromosome-level genome assembly of Paracoccus marginatus based on PacBio and Hi-C technologies. Sci Data 12, 901 (2025). https://doi.org/10.1038/s41597-025-04944-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04944-7
