
Abstract
Bryophytes play a crucial role in the ecosystems of polar regions. These simple plants are among the predominant vegetation types in both Arctic and Antarctic landscapes, where they contribute significantly to biodiversity and ecological stability. Here, we report the chromosome-level genomes of two polar moss species, the Arctic Aulacomnium turgidum and Antarctic Polytrichastrum alpinum. Utilizing a combination of Illumina short reads, Nanopore long reads, and Hi-C data, we assembled genomes of 277.84 Mb for A. turgidum and 498.33 Mb for P. alpinum, respectively. These assemblies were anchored to 11 chromosomes for A. turgidum and 8 chromosomes for P. alpinum. Both species exhibited a sex chromosome with distinct genomic characteristics. Gene annotations revealed 25,999 protein-coding genes in A. turgidum and 28,070 in P. alpinum. The high completeness of the gene space was validated via BUSCO, achieving impressive scores of 98.2% and 98.0%. These high-quality genomes provide critical resources for studying the adaptive evolution and stress tolerance mechanisms of mosses in extreme polar environments.
Similar content being viewed by others


Background & Summary
Bryophytes, including hornworts, liverworts, and mosses, form the sister group to vascular plants, and exhibit a haploid-dominated lifecycle with unbranched sporophytes remain attaching to and reliant on gametophytes1. Bryophytes excel in enduring some of the most severe environmental conditions, ranging from low light intensity and extreme temperatures to nutrient scarcity and desiccation. As a result, they often act as pioneering species within various ecosystems. Notably, bryophytes are the predominant flora in Antarctica’s terrestrial ecosystems2 and can also thrive in arid deserts3. These adaptabilities may stem from an evolutionarily refined genetic toolkit geared towards stress tolerance4.
Aulacomnium turgidum (Aulacomniaceae, Rhizogoniales), a moss species widespread across the Arctic Circle, including Svalbard, Greenland, and Alaska, is notable for its ability to regenerate after being entombed in ice for 400 years, demonstrating exceptional freeze stress tolerance5. The A. turgidum (Fig. 1a) was collected from Svalbard, a Norwegian archipelago, epitomizes a typical Arctic environment. The annual mean surface air temperatures vary from 2.5 to 5.8 °C in summer and to −11.4 to −9.4 °C in winter6. Despite these extreme conditions, resilient plants, including bryophytes, flourish due to their high tolerance for abiotic stresses7,8.

(a) Aulacomnium turgidum and (b) geographical map showing its sampling site in the Arctic. (c) Polytrichastrum alpinum and (d) geographical map showing its sampling site in the Antarctic.
Polytrichastrum alpinum (Polytrichaceae, Polytrichales) is a common Antarctic moss species, and thrives in moist, rocky environments near glacier moraine peaks, as well as in dry areas2. The P. alpinum sample (Fig. 1c) was collected from King George Island, located at the northern tip of the Antarctic Peninsula, an area characterized by its semi-desert landscape9. The average annual temperature of this area is −2.3 °C, with summer months experiencing temperatures slightly above freezing. The region is subject to frequent strong winds10. The island is home to 64 documented species of moss, which are primarily found in humid, sheltered areas with relatively stable and partially organic soils11,12.
Over the recent decades, research has utilized mosses including A. turgidum and P. alpinum from the Arctic and Antarctic regions to study the impacts of global warming on Earth13,14, the interactions between polar microbial communities and plants15,16, and the molecular mechanisms behind unique survival strategies in extreme conditions17,18. Despite the ecological significance of polar mosses, our understanding of their genomes remains limited19. These mosses, which thrive under the harsh conditions of the Arctic and Antarctic, are critical for understanding resilience and adaptability to extreme environments. However, the scarcity of complete genomic data hinders our ability to fully comprehend the molecular mechanisms that underpin their unique survival strategies. This gap in knowledge underscores the need for enhanced genomic research to better exploit the potential of polar mosses in studying environmental adaptation and climate change resilience.
In this study, we assembled high-quality chromosome-level genomes of A. turgidum and P. alpinum using a combination of Illumina short reads, Nanopore long reads, and high-throughput chromosome conformation capture (Hi-C) data. The genome of A. turgidum was assembled as 277.84 Mb with a contig N50 of 11.92 Mb, while the genome of P. alpinum was 498.33 Mb with a contig N50 of 4.24 Mb (Table 1). A total of 275.60 Mb and 488.51 Mb of the assemblies were anchored to 11 and 8 chromosomes for A. turgidum and P. alpinum, respectively (Table 2). Both species possess a sex chromosome with lower gene density and higher repetitive sequence density than the autosomes. A. turgidum and P. alpinum respectively encodes 25,999 and 28,070 protein-coding genes (Table 3). These two high-quality genomes offer valuable new genomic resources for future research into the genetic foundations and adaptive evolution of plants in Arctic and Antarctic environments.
Methods
Plant materials and sequencing
Wild gametophytes of A. turgidum were collected from Spitsbergen Island, Svalbard (78°54′41″ N, 11°58′35″ E) (Fig. 1b) on September 18th, 2018, and P. alpinum from King George Island, Antarctica (62°12.041” S, 58°59.698” W) (Fig. 1d) on December 23rd, 2018. Voucher specimens were deposited in the Herbarium of Shenzhen Fairy Lake Botanical Garden in Shenzhen, China (SZG). The entire moss plant, including its leaves, stems, and rhizoids, was used for sequencing. Approximately 5 grams of material were gathered from these samples for genomic and transcriptomic sequencing, respectively. Three distinct types of genome sequencing techniques were performed: the Nanopore long-read sequencing, the Hi-C sequencing, and the Illumina short-read sequencing. Additionally, plant tissues were utilized for transcriptomic sequencing. Transcriptome libraries were constructed using a TruSeq RNA Library Prep Kit v2 (Illumina, CA, USA), with an insert size ranging from 200 to 400 bp, following polyA selection. Sequencing was performed on an Illumina NovaSeq 6000 platform (Illumina, CA, USA), generating 150-bp paired-end reads.
Genome assembly
Illumina short-read sequencing and genome survey
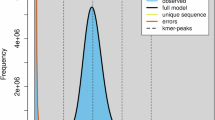
The Illumina short reads were processed using Trimmomatic (v0.39)20 to filter out duplicates, low-quality reads, and adapters. Filtered reads were used for k-mer analyses to estimate genome sizes. Jellyfish (v2.0.0)21 was employed to count the k-mer frequencies with the ‘-m 21’ parameter. GenomeScope (v2.0)22 was used to estimate genome sizes with the settings “-k 21 -p 1” along with other default parameters. These analyses predicted that the genome sizes for A. turgidum and P. alpinum are 272.00 Mb and 449.50 Mb, respectively, with the proportion of repetitive sequences at 29.52% and 40.00%, respectively (Fig. 2).

Genome survey based on k-mer (k = 21) analysis of A. turgidum and P. alpinum. The X-axis represents the k-mer depth, while the Y-axis indicates the k-mer frequency for a given depth.
ONT long-read sequencing and genome assembly
Nanopore libraries were prepared using SQK-LSK108 and sequenced on a Nanopore PromethION sequencer. The long reads obtained were assembled using NextDenovo (v2.5.0)23. The initial assembly was then polished for three iteratives with NextPolish (v1.3.1)24, using both Nanopore long reads and filtered Illumina reads.
Hi-C scaffolding and chromosome-level genome assembly
The Hi-C library construction involved several steps: cross-linking, restriction enzyme digestion (using MboI), end repair, DNA cyclization, and purification25. Paired-end 150-bp reads were generated on an Illumina NovaSeq 6000 platform (Illumina, CA, USA). The raw Hi-C reads were initially processed with Trimmomatic (v0.39)20 using default parameters. Subsequently, Juicer (v1.6)26 was employed to extract valid data. Misassembled contigs were corrected, anchored, ordered, and oriented using the 3D-DNA pipeline (v 180922)27. Juicebox (v1.11.08)28 was then utilized for manual adjustments of the anchored results. For scaffolds not anchored to chromosomes, we performed the procedure outlined by Zhou et al.29 to remove those containing contamination or organelle fragments, and to link consecutive contigs to generate a high-quality genome assembly (Fig. 3). This process ultimately enabled the identification of 11 chromosomes for A. turgidum and 8 chromosomes for P. alpinum, resulting in final chromosome-scale genome assemblies of 277.84 Mb for A. turgidum and 498.33 Mb for P. alpinum (Table 1).

Hi-C interaction heatmap for A. turgidum and P. alpinum. The heatmap illustrates the density of Hi-C interactions among distinct chromosomes, showing scaffolded and independently assembled chromosomes at high resolution. The intensity of red color reflects to the number of Hi-C interactions.
Repeat annotation
For repeat annotation, a customized de novo repeat library was created using a homology-based approach. Programs Piler (v0.4.1)30, LTR_FINDER (v1.0.5)31, RepeatScout (v1.0.5)32, and RepeatModeler (v2.0)33 were used to generate the libraries. The resulting repetitive sequence libraries were combined and used as input for RepeatMasker (v.4.1.1)34. Additionally, Repbase (v21.01)35 served as the database for known repetitive elements, searched using RepeatMasker (v.4.1.1)34. Tandem Repeats Finder (TRF v4.07)36 was employed to predict tandem repeat sequences across the genome. The results indicated that the genomes of A. turgidum and P. alpinum contained 42.65% and 53.37% repetitive elements, respectively. The percentages of long terminal repeat (LTR) retrotransposons were 15.45% for A. turgidum and 25.16% for P. alpinum (Table 3 and Fig. 4).

Circos plots illustrating the chromosomal-level genome assemblies for A. turgidum (left) and P. alpinum (right). The tracks, from outermost to innermost, are: chromosomes, GC content, gene density, TE density, LTR/Gypsy density, LTR/Copia density and color ribbons representing genome-wide syntenic blocks of inter-chromosome and intra-chromosome. The color bar indicates the proportion of GC content.
Gene and functional annotation
To predict protein-coding genes (PCGs) for A. turgidum and P. alpinum, we integrated two types of evidence. We sourced proteome sequences from Physcomitrium patens37, Marchantia polymorpha (https://marchantia.info/), Arabidopsis thaliana (https://www.arabidopsis.org/), and the bryophyte proteins from Swiss-Prot (https://www.uniprot.org/) to provide homology-based protein evidence. Transcriptome evidence involved mapping clean illumina reads to the assembled genomes using HISAT (v2.2.0)38 to produce BAM files. Gene models were then predicted using BRAKER3 pipeline (v3.0.6)39, in conjunction with AUGUSTUS40 and GeneMark41 de novo predictions, based on the soft-masked genome, protein evidence, and transcriptome data. Additionally, transcripts were generated using StringTie (v2.2.1)42 and TransDecoder (v5.5.0) (https://github.com/TransDecoder/TransDecoder). These pieces of evidences were integrated using EVidenceModeler (v2.1.0)43. Subsequently, Trinity (v2.8.4)44 was used for de novo transcript assembly, updating gene models to include untranslated regions (UTRs) and alternative splicing variants via the PASA pipeline (v2.5.3)45. A total of 25,999 and 28,070 protein-coding genes were predicted in A. turgidum and P. alpinum, respectively (Table 3). The completeness of the gene space was evaluated using the BUSCO v.3.1.0, based on the Viridiplantae_odb10 set46. For functional annotation, gene models were aligned against the UniProt (Swiss-Prot and TrEMBL), KEGG, and TAIR databases. Protein domains and gene ontologies were annotated using InterProScan (v5.51–85.0)47. Additionally, the iTAK online tool (v1.6)48 was used to identify transcription factors, predicting 727 transcription factors in A. turgidum and 698 in P. alpinum, (Table 4, Tables S1, S2).
Non-coding RNA annotation
Transfer RNAs (tRNAs) were identified using tRNAscan-SE (v1.3.1)49 with default parameters. Given the high conservation of ribosomal RNAs (rRNAs), rRNA sequences from closely related species were downloaded from the Ensembl database and used as references in BLAST (v2.2.26)50 search with an e-value threshold of 1e-5. Other non-coding RNAs (ncRNAs), including micro RNAs (miRNAs) and small nuclear RNAs (snRNAs), were identified by searching against the Rfam database (v12.0)51 using Infernal (v1.1.1)52 with default parameters. In total, 1,032 and 3,223 ncRNAs were identified in A. turgidum and P. alpinum, respectively (Table 5).
Genome synteny analysis and Circos diagram construction
The python library jcvi (v1.1.8)53 was employed to identify intra-genomic syntenic blocks and detect inter- and intra-chromosomal synteny (Table S3). The syntenic regions were first identified using the jcvi.compara.catalog ortholog function with a default C-score threshold of 0.7 to filter out low-quality hits. Syntenic depths were then calculated using the jcvi.compara.synteny depth function. To further refine the syntenic blocks, we applied the jcvi.compara.synteny screen function with the parameters–minspan = 30–simple. Visualization of syntenic relationships and genomic feature distributions was carried out using Circos54. The Circos plot (Fig. 4) includes synteny blocks, GC content, gene density, transposable element (TE) density, LTR/Gypsy density, and LTR/Copia density, with all features calculated in 1 Mb windows.
Phylogenetic reconstruction
A total of 14 bryophyte species, including the newly sequenced A. turgidum and P. alpinum, were sampled to identify orthologs using OrthoFinder (v2.3.11)55. Alignment for protein sequences of 61 single-copy orthologs was performed using MAFFT (v7.453)56. The resulting alignments were then concatenated and used as the input data for IQ-TREE2 (v2.0.6)57 to construct a maximum likelihood phylogenetic tree, with the JTT model and 1,000 ultrafast bootstrap replicates. The resulting phylogenetic tree was rooted with Anthoceros angustus, and visualized using the interactive Tree of Life (iTOL)58 (Fig. 5).

A maximum likelihood phylogenetic tree of bryophytes based on 61 single-copy ortholog protein sequences from 14 bryophyte species. The newly sequenced two mosses are highlighted in bold.
Data Records
The raw data from Nanopore, Hi-C, Illumina short-read sequencing used for genome assembly and annotation have been deposited in the Genome Sequence Archive (GSA) of the National Genomics Data Center (NGDC) with the accession number CRA01759659 under the BioProject accession number PRJCA02776060. All the genomic sequencing raw data were also deposited in the China National GeneBank Database (CNGB) Nucleotide Sequence Archive (CNSA) under accession numbers CNP000289561. The final contigs and chromosome assemblies are submitted to NCBI under the accession number GCA_048933245.162, GCA_048933195.163 of A. turgidum and P. alpinum, respectively. The contigs and chromosome-scale genome assemblies have also been made available in the GSA at the NGDC. The specific accession numbers for sequences of A. turgidum and P. alpinum are GWHEUUP00000000.164 and GWHEUUQ00000000.165, respectively. The annotation files are available in figshare66.
Technical Validation
The completeness and accuracy of the genome assemblies for A. turgidum and P. alpinum were assessed using multiple approaches. First, Benchmarking Universal Single-Copy Orthologs (BUSCO)46 analyses with the Viridiplantae_odb10 indicated completeness scores of 98.20% for A. turgidum and 98.00% for P. alpinum. Additionally, QUAST67 was employed to evaluate assembly contiguity metrics, revealing final assembly size of 277.84 Mb for A. turgidum with a L50 value of 5, and 498.33 Mb for P. alpinum with a L50 value of 3. To further assess assembly accuracy, Merqury68 was used, yielding quality values (QV) of 30.14 for A. turgidum and 29.59 for P. alpinum, with assembly completeness rates of 99.42% and 93.06%, respectively. The interaction contact pattern, centered along the principal diagonal in the Hi-C heatmap (Fig. 3), further supports the accuracy of the chromosome -level assemblies.
Code availability
All codes and pipelines used for data processing was executed following the manual and protocols of the respective bioinformatic tools. Detailed information on the software versions is provided in the Methods section. No custom code was developed for this study.
References
Goffinet, B. & Buck, W. R. The evolution of body form in bryophytes. Annual Plant Reviews, 51–89 (2013).
Ochyra, R., Bednarek-Ochyra, H. & Smith, R. I. L. The Illustrated Moss Flora Of Antarctica (Cambridge University Press, 2008).
Pan, Z. et al. The upside-down water collection system of Syntrichia caninervis. Nature Plants 2, 16076 (2016).
Kulshrestha, S. et al. Stress, senescence, and specialized metabolites in bryophytes. Journal of Experimental Botany 73, 4396–4411 (2022).
La Farge, C., Williams, K. H. & England, J. H. Regeneration of little ice age bryophytes emerging from a polar glacier with implications of totipotency in extreme environments. Proceedings of the National Academy of Sciences 110, 9839–9844 (2013).
Maturilli, M., Herber, A. & König-Langlo, G. Climatology and time series of surface meteorology in Ny-Alesund, Svalbard. Earth System Science Data 5, 155–163 (2013).
Crawford, R. M. M., Chapman, H. M. & Hodge, H. Anoxia tolerance in high arctic vegetation. Arctic and Alpine Research 26, 308 (1994).
Liu, S. et al. Complementary DNA library construction and expressed sequence tag analysis of an arctic moss, Aulacomnium turgidum. Polar Biology 33, 617–626 (2010).
Rakusa-Suszczewski, S. King George Island — South Shetland Islands, Maritime Antarctic (Springer Berlin Heidelberg, 2002).
Oliva, M. et al. Recent regional climate cooling on the antarctic peninsula and associated impacts on the cryosphere. Science of The Total Environment 580, 210–223 (2017).
Ochyra, R. The Moss Flora Of King George Island, Antarctica (Polish Academy of Sciences, 1998).
Li, S. et al. Drepanocladus longifolius (Amblystegiaceae), an addition to the moss flora of king george island, south shetland islands, with a review of antarctic benthic mosses. Polar Biology 32, 1415–1425 (2009).
Shortlidge, E. E. et al. Passive warming reduces stress and shifts reproductive effort in the antarctic moss, Polytrichastrum alpinum. Annals of Botany 119, 27–38 (2017).
Koncz, P., Hermanutz, L., Marino, P., Wheeler, J. & Cranston, B. Bryophyte community diversities and expected change under a warming climate in contrasting habitats of the Torngat Mountains, Labrador. The Bryologist 121, 174–182 (2018). 179.
Koranda, M., Rinnan, R. & Michelsen, A. Close coupling of plant functional types with soil microbial community composition drives soil carbon and nutrient cycling in tundra heath. Plant and Soil 488, 551–572 (2023).
de Carvalho, C. R. et al. Cultivable fungi associated with bryosphere of bipolar mosses Polytrichastrum alpinum and Polytrichum juniperinum in King George Island, South Shetland Islands, Maritime Antarctica. Polar Biology 43, 545–553 (2020).
Alavilli, H., Lee, H., Park, M., Yun, D. J. & Lee, B. H. Enhanced multiple stress tolerance in Arabidopsis by overexpression of the polar moss peptidyl prolyl isomerase FKBP12 gene. Plant Cell Reports 37, 453–465 (2018).
Kang, P., Yoo, Y.-H., Kim, D.-I., Yim, J. H. & Lee, H. De novo transcriptome assembly and comparative analysis of differentially expressed genes involved in cold acclimation and freezing tolerance of the arctic moss Aulacomnium turgidum (Wahlenb.) Schwaegr. Plants 12, 1250 (2023).
Liu, S. et al. The antarctic moss Pohlia nutans genome provides insights into the evolution of bryophytes and the adaptation to extreme terrestrial habitats. Frontiers in Plant Science 13, 920138 (2022).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432 (2020).
Hu, J. et al. NextDenovo: an efficient error correction and accurate assembly tool for noisy long reads. Genome Biology 25, 107 (2024).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2019).
Yu, J. et al. Chromosome-Level genome assemblies of two Hypnales (mosses) reveal high intergeneric synteny. Genome Biology and Evolution 14 (2022).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems 3, 99–101 (2016).
Zhou, X. et al. Chromosome-level genome assembly of Niphotrichum japonicum provides new insights into heat stress responses in mosses. Frontiers in Plant Science 14 (2023).
Edgar, R. C. & Myers, E. W. PILER: identification and classification of genomic repeats. Bioinformatics 21, 152–158 (2005).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21(Suppl 1), i351–358 (2005).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences of the United States of America 117, 9451–9457 (2020).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics 25, 4.10.11–14.10.14 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Bi, G. et al. Near telomere-to-telomere genome of the model plant Physcomitrium patens. Nature Plants 10, 327–343 (2024).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nature Biotechnology 37, 907–915 (2019).
Gabriel, L. et al. BRAKER3: fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Research 34, 769–777 (2024).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–439 (2006).
Bruna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics 2 (2020).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature Biotechnology 33, 290–295 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology 9, R7 (2008).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nature Protocols 8, 1494–1512 (2013).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31, 5654–5666 (2003).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral Genomes. Molecular Biology and Evolution 38, 4647–4654 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Zheng, Y. et al. iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Molecular Plant 9, 1667–1670 (2016).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research 25, 955–964 (1997).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 25, 3389–3402 (1997).
Nawrocki, E. P. et al. Rfam 12.0: updates to the RNA families database. Nucleic Acids Research 43, D130–D137 (2014).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Tang, H. et al. JCVI: a versatile toolkit for comparative genomics analysis. Imeta 3, e211 (2024).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Research 19, 1639–1645 (2009).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology 20, 238 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30, 772–780 (2013).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution 37, 1530–1534 (2020).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 (2006).
NGDC Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA017596 (2025).
NGDC BioProject https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA027760 (2025).
CNGB Nucleotide Sequence Archive https://db.cngb.org/search/project/CNP0002895 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_048933245.1 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_048933195.1 (2025).
NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/85929/show (2025).
NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/85930/show (2025).
Zeng, Y. Y. The annotation files of Aulacomnium turgidum and Polytrichastrum alpinum. figshare. https://doi.org/10.6084/m9.figshare.28595150 (2025).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21, 245 (2020).
Acknowledgements
This study was supported by the 10KP project (https://db.cngb.org/10kp/) and China National GeneBank (CNGB; https://www.cngb.org/). Additionally, this study was supported by the Scientific Foundation of the Urban Management Bureau of Shenzhen (202005, 202403 to Y.L., and 202106, 202302 to S.D.).
Author information
Authors and Affiliations
Contributions
Y.L., S.D., H.Y. and T.W. conceived the study. Y.Y. collected the materials. Y.Z., Y.C., X.Z., J.Y. and X.L. performed the analyses. Y.Z. drafted the manuscript. All authors read and approved the manuscript. Y.Z. and Y.C. contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zeng, Y., Cai, Y., Zhou, X. et al. Chromosome-level genomes of Arctic and Antarctic mosses: Aulacomnium turgidum and Polytrichastrum alpinum. Sci Data 12, 705 (2025). https://doi.org/10.1038/s41597-025-04960-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-04960-7
