
Abstract
The rhizosphere microbiota recruited by plants contributes significantly to maintaining host productivity and resisting stress. However, the genetic mechanisms by which plants regulate this recruitment process remain largely unclear. Here, we generated a comprehensive dataset, including 27 root transcriptomes, 27 root metabolomes, and 54 bulk or rhizosphere soil 16S rRNA amplicons across nine poplar species from four sections grown in nutrient-poor natural soil, along with eleven growth phenotype data. We provided a thorough description of this dataset, followed by a comprehensive co-expression network analysis example that broke down the wall of the four-way relationship between plant gene-metabolite-microbe-phenotype, thus identifying the links between plant gene expression, metabolite accumulation, growth behavior, and rhizosphere microbiome variation under nutrient-poor conditions. Overall, this dataset enhances our understanding of plant and microbe interactions, offering valuable strategies and novel insights for resolving how plants regulate rhizosphere microbial compositions and functions, thereby improving host fitness, which will benefit future research.
Similar content being viewed by others


Background & Summary
By shaping the composition and functionality of the rhizosphere microbial community and maintaining its dynamic equilibrium, plants achieve a good synergy with microorganisms, which dynamically affects plant growth, and resistance to biological and abiotic stresses1,2,3. Distinct plant genotypes often harbor unique microbiomes, and their microbial composition even mirrors host phylogeny4,5. The microbial composition and diversity within the endosphere of 30 plant species that have diverged for up to 140 million years were related to plant phylogeny6. Evidently, it is host gene expression that plays a selective role. The functions of the plant genes themselves are crucial for understanding the signaling cascades that control plant development and stress responses7,8. Concurrently, the microbial taxa recruited by these genes also contribute significantly to plant fitness9,10,11. However, how the plant rhizosphere microbiota is recruited during establishment and what role genes play in this process remain largely unclear.
Plants indirectly drive the aggregation of rhizosphere microbial communities by secreting specific metabolites, such as flavonoids, benzoxazinoids, and coumarins11,12,13. Flavonoids are the most extensively studied class of such secretions and are recognized as crucial root-released rhizosphere signal molecules that modulate plant-microbe interactions3,14. In legumes, root flavonoids were known to play a crucial role in the formation of root nodules during symbiosis with diazotrophic bacteria15. Various types of flavonoids, including flavones, flavonols, flavanones, isoflavones, and chalcones, serve as plant signals that induce the expression of nod genes in rhizobia16,17,18. Recent research indicates that the activity of non-nodular bacteria associated with non-legumes under stress was also affected by flavonoids. For example, flavonoid biosynthesis (apigenin) in rice enhances the biofilm formation of Gluconacetobacter diazotrophicus and the biological nitrogen fixation of soil diazotrophic bacteria19; flavonoid (naringenin) enhances the dehydration resistance of Arabidopsis by enhancing the Aeromonas sp. H1 motility and colonization20. Therefore, elucidating the causal relationship between flavonoids and beneficial microbes is critical and challenging. The establishment of a comprehensive dataset and network involving genes, flavonoids, and rhizosphere microbes is necessary to understand the molecular mechanisms of rhizosphere microbial recruitment.
Poplar (Populus L.), as a model woody perennial plant, is categorized into five sections: Leuce, Aigeiros, Tacamahaca, Turanga, and Leucoides21. Researches have revealed that diverse poplar genetic populations have different affinity and efficiency for nitrogen and phosphorus uptake22, and shape the respective rhizosphere microbiome by recruiting specific microbial taxa23,24,25, under which microbes may alter host performance and fitness directly or via ecosystem services such as nutrient accessibility9,26. In-depth exploration of the intricate interplay between poplar and microorganisms under abiotic stress holds promise for cultivating novel germplasm resources, which provides a new direction for genetic breeding.
Here, we present an integrated dataset which contains root transcriptome, root metabolome, rhizosphere microbiome, and phenotypes from nine representative poplar species of four sections under nutrient-poor conditions (total nitrogen: 0.089%; available phosphorus: 12.4 mg/kg; available potassium: 158.6 mg/kg; organic matter: 22.6 g/kg), which enables precise profiling of the activities of host genes regulating metabolites mediating microbial composition and their interactions with plant phenotypes. We examined eleven representative characteristics encompassing morphological and structural, physiological functional, and component content aspects of all poplar species. Moreover, a total of 73.7 Gb of root transcriptomic raw data was generated, which, after quality filtering, retained 70.0 Gb of clean data and identified 38,739 expressed genes (TPM > 0). Bacterial community composition across the nine poplar species was investigated for each sample type (bulk soil and rhizosphere soil) using Illumina MiSeq sequencing of the V3-V4 region of the 16S rRNA gene. The 54 soil samples identified 30,225 absolute sequence variants (ASVs). We detected and quantified 129 root flavonoids representing diverse chemical classes: 49 flavones, 30 flavonols, 14 flavanones, 7 anthocyanins, 11 isoflavones, 5 dihydroflavonols, and 13 other flavonoids.
This combined dataset can provide a comprehensive multi-omics analysis of plant-microbe interactions, potentially play an important role in further investigations into the mechanisms of specific genes, metabolites, and microbes in these interactions, as well as their impact on plant growth and development under nutrient deficiency conditions, and offer a valuable basis for studying the effects of plant-metabolite-microbial regulatory models on poplar adaptation.
Methods
Plant and soil materials, growth conditions
Nine species of poplars from four sections (Table 1), Leuce (Populus tomentosa × P. bolleana M: Pto-M, P. alba × P. glandulosa 84 K: 84 K, P. alba × P. glandulosa Y: Pal-Y, P. tomentosa ‘Lumao50’: LM50), Aigeiros (P. euramericana ‘74/76’: 107, P. euramericana H3-1: H3-1), Tacamahaca (P. trichocarpa M: Pot-M, P. szechuanica Z: Psz-Z), and Turanga (P. euphratica H: Peu-H), were examined in this study. The 84 K, Psz-Z, Pot-M, and Peu-H poplar clones were maintained in our laboratory, while the remaining five species were collected from the GuanXian state-owned P. tomentosa forest farm in Shandong Province, China (E: 115°22′8″, N: 36°30′54″) to obtain sterile monoclonal tissue culture seedlings.
The natural soils of Leuce, Aigeiros, and Tacamahaca were collected from P. tomentosa, P. euramericana, and P. simonii plantations in the GuanXian state-owned forest farm, while Turanga soil was obtained from the natural P. euphratica forest in the Danglang tribe, Aksu, Xinjiang (E: 80°15′18″, N: 40°45′39″). Notably, the poplars, all over 15 years old, had never been fertilized. Soil was collected at a depth of 10–40 cm after removing the top 10 cm from a radius of 2–3 m around each poplar. Each forest soil sample was pooled from at least five trees and mixed.
To provide more abundant microbes for different genotypes of poplar, equal volumes of four natural soils were mixed thoroughly. Tissue culture seedlings of the nine poplar species were then transplanted into the mixed soil (total nitrogen: 0.089%) for pot experiments, with at least five biological replications per species. The seedlings were randomly arranged and grown under identical conditions (25 °C; 16 h day/8 h night light cycle) for three months, watered every two days. To support normal growth, each pot received 300 ml of a 1 g/L fertilizer solution (Huaduo 1, China; 20% N, 20% P, 20% K) after transplanting, with no further fertilization applied.
Plant measurements and sample collection
On the day of destructive sampling, eleven representative traits (P-values < 0.01; Fig. 1) were measured across nine poplar species, covering morphological and structural (growth traits: plant height, ground diameter, shoot biomass, root biomass, and root length; leaf traits: leaf length, leaf width, leaf area, and leaf number), physiological functional (chlorophyll content), and component content (leaf nitrogen content). Due to insufficient root and rhizosphere soil from one Peu-H plant for subsequent sequencing, two plant samples were combined as one biological replicate, and their phenotypic data averaged. For other species, three plants with similar growth were selected as biological replicates. Leaf length and width were measured for the third to fifth fully expanded leaves, and leaf area was calculated using ImageJ software27 (v.1.53q). Fully expanded leaves were defined as those exceeding 4.0 cm in length. Chlorophyll content was averaged from 20 readings taken with a SPAD-502 Plus meter (Konica Minolta, Japan) in the middle third of the leaf in the longitudinal direction. Above-ground and below-ground biomass of poplar was harvested, and fresh biomass was determined. Leaves from the 3rd, 4th, and 5th nodes from the top were collected for each species, thoroughly dried at 60 °C, and finely ground into powder. The nitrogen content of the leaves was measured using the Elementar Vario PYRO Cube elemental analyzer (Vario PYRO Cube, Germany). The soil chemical traits (total nitrogen, available phosphorus, and soil organic matter) were measured using a previously described method28. Available potassium was analyzed by the flame photometer method29. One-way ANOVA was used to assess significant differences in the data.

Phenotypic differences of nine poplar species from four sections. (a) Diversity in leaves between nine poplar species (the third leaf completely expanded at the top). a: Pto-M; b: 84 K; c: LM50; d: Pal-Y; e: H3-1; f: 107; g: Psz-Z; h: Pot-M; i: Peu-H. Fresh shoot biomass (b), fresh root biomass (c), root length (d), leaf area (e), and leaf nitrogen content (f) of four poplar sections. Different letters indicate significantly different groups (One-way ANOVA, P-values < 0.05). FW, fresh weight. Scale bars: (a) 5 cm.
The rooting system of each plant was subsampled to assess various response variables: root asmples for flavonoids metabolite analysis and root transcriptome analysis, and rhizosphere soils for 16S rRNA amplicon-based sequencing (Fig. 2). To ensure the focus was on the most active roots30, only fine roots (<2 mm in diameter) were utilized for these analyses. For metabolomics and transcriptomics, roots were quickly rinsed with deionized water and immediately frozen in liquid nitrogen. Rhizosphere soil samples were collected by placing fine roots into a 50-ml centrifuge tube containing 30 ml of sterile Phosphate Buffer Saline (PBS, pH 7.0; per liter: 6.33 g NaH2PO4·H2O, 16.5 g Na2HPO4·7H2O, and 200 μl Silwet L-77), and stored on ice for further processing. Bulk soil samples from around the root system were also immediately frozen in liquid nitrogen. Rhizosphere samples were extracted from the corresponding root segments, following previous protocols with minor modifications31. Centrifuge tubes with samples were shaken for 30 min at 50 rpm in a constant-temperature shaker incubaror, with the process repeated twice. Samples were centrifuged for 10 minutes at 4,000 rpm at 4 °C, the supernatant removed, and the rhizosphere soil resuspended in sterile water before being frozen in liquid nitrogen and stored at −80 °C until further analysis.

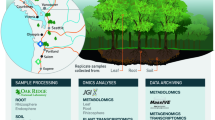
Schematic overview of the experimental pipeline. Fine-root (< 2 mm), rhizosphere soil, and bulk soil samples were collected from nine poplar species of four sections growing in low nitrogen natural soil, three biological replicates per species. The samples were quickly placed in liquid nitrogen and stored at −80 °C. Transcriptome and flavonoid metabolites sequencing were performed for root samples, 16S rRNA amplicon sequencing was performed for rhizosphere soil and bulk soil samples. Multi-omics analyses were performed on all data to gain detailed insights into the mechanisms of gene-metabolite-microbe interactions, and the dataset was made available for further research. The methods or other conditions for each step are listed below the steps. For further details, please see the Methods of the manuscript.
16S rRNA gene sequencing and microbiome analysis
16S rRNA gene sequencing. The rhizosphere and bulk soil samples were fully ground in liquid nitrogen, and genomic DNA was extracted using the CTAB method described previously32. Specific regions of the 16S rRNA gene (V3-V4) were amplified with barcode-labeled primers (V3-V4; 341 F: CCTAYGGGRBGCASCAG, 806 R: GGACTACNNGGGTATCTAAT). Each PCR reaction contained 15 μl of Phusion® High-Fidelity PCR Master Mix (New England Biolabs, USA), 0.2 μM of forward and reverse primers, and around 10 ng of template DNA. The resulting PCR products were purified using a Gel Extraction Kit (Qiagen, Germany).
Sequencing libraries were constructed using the TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA) following the manufacturer’s protocol, with index codes added. Library quality was evaluated with the [email protected] Fluorometer (Thermo Scientific, USA) and the Agilent Bioanalyzer 2100 system. Finally, the libraries were sequenced on the Illumina NovaSeq platform, producing 250 bp paired-end reads.
Amplicon sequence data analysis. The 16S rRNA gene sequences were processed using QIIME33 (v.1.9.1), USEARCH34 (v.10.0) and in-house scripts. Paired-end read quality was assessed via FastQC35 (v.0.11.5) and subsequently processed with USEARCH: joining of paired-end reads and relabeling of sequencing names (-fastq_mergepairs); removal of barcodes and primers (-fastx_truncate); filtering of low-quality reads (-fastq_filter); and finding non-redundancy reads (-fastx_uniques). Unoise3, with default parameters36, was used to cluster reads at 100% sequence similarity. Taxonomic assignment utilized the SILVA reference database (v132) for bacteria37. Separate tables were generated for each taxonomic level (Domain to Genus)38,39. All ASVs identified as chloroplast and mitochondria were discarded from the dataset, and ASVs represented by fewer than two sequences were filtered to avoid biases. QIIME (v.1.9.1) was used to calculate the alpha diversity (Shannon, Chao1) and beta diversity (weighted/unweighted Unifrac and Bray-Curtis dissimilarity) indices for microbial community analysis across soil samples.
Root total RNA extraction, transcriptome sequencing and analysis
Root total RNA extraction and transcriptome sequencing. Total RNA was extracted from root tissues of nine poplar species. The frozen roots were fully ground in liquid nitrogen, and RNA was extracted using the RNeasy Mini Kit (Qiagen, Germany) as per the manufacturer’s protocol. RNA quality was confirmed by agarose gel electrophoresis, ensuring the presence of distinct, unambiguous bands. Concentration and 260/280 nm ratios were measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). RNA-seq cDNA libraries were constructed using samples that met rigorous quality criteria, and the cDNA libraries were synthesized using the GoScript™ Reverse Transcription System (Promega, USA). Sequencing was performed on the HiSeq 6000 platform (Illumina, USA), acquiring 150 bp paired-end reads.
Transcriptome analysis. Transcriptome profiling was performed as established protocols40. In brief, raw RNA-seq data underwent stringent quality control using fastp (v.0.14.0)41. Paired reads possessing adapters, exhibiting an N content < 10% or with > 50% low-quality nucleotides (sQ ≤ 5) were methodically eliminated from the dataset. Clean reads obtained from the Hiseq X Ten sequencing platform were mapped to the P. trichocarpa reference genome42 (v.3.1; https://phytozome-next.jgi.doe.gov/info/Ptrichocarpa_v3_1; Phytozome genome ID: 444) using HISAT2 (v.2-2.1.0)43. The resulting sam files were converted to bam files, sorted with samtools (v.0.1.19), and normalized to TPM using StringTie (TPM > 0; v.1.3.6)44. Qualimap (v.2.2.1)45 was used to evaluate the quality of transcriptome data alignment. Differentially expressed genes were analyzed with DESeq 2 (v.1.34.0; |log2FC| ≥ 1, FDR adjusted P-values < 0.05).
Metabolites measurement and metabolome analysis
Metabolites extraction
Fine-root samples (< 2 mm diameter; 100 mg) were finely ground in liquid nitrogen, and the homogenate was mixed with prechilled 80% MeOH and 0.1% formic acid via vortexing. The mixtures were centrifuged at 10,000 g at 4 °C for 20 min. A portion of the supernatant was diluted to achieve a final 53% MeOH concentration. These processed samples were transferred to new tubes and centrifuged again at 10,000 g at 4 °C for 20 min. The final supernatants were subjected to LC-MS/MS system for analysis. In order to ensure instrument stability, the samples were equally amalgamated to form multiple quality control (QC) samples.
HPLC-MS/MS analysis
LC-MS/MS analyses were conducted using the ExionLC™ AD system (SCIEX, USA) connected to a QTRAP® 6500 + mass spectrometer (SCIEX, USA). Samples were injected onto an Xselect HSS T3 column (2.1 × 150 mm, 2.5 μm) with a 20-min linear gradient at a 0.4 ml/min flow rate. Eluent A was 0.1% formic acid-water, and eluent B was 0.1% formic acid-acetonitrile. The solvent gradient was: 2% B, 2.0 min; 2–100% B, 15.0 min; 100% B, 17.0 min; 100-2% B, 17.1 min; 2% B, 20.0 min. The QTRAP® 6500 + mass spectrometer operated in positive polarity mode with an IonSpray voltage of 5500 V and a temperature of 550 °C or in negative polarity mode with an IonSpray voltage of −4500 V and a temperature of 550 °C.
Metabolites identification and quantification
Sample analysis using Multiple Reaction Monitoring (MRM) was conducted based on Novogene’s proprietary database and FlavonoidSearch46. Metabolite quantification relies on Q3 for accurate measurements. Metabolite identification utilized Q1, Q3, retention time (RT), declustering potential (DP), and collision energy (CE). Data files generated by HPLC-MS/MS were processed with SCIEX OS (v.1.4) for peak integration and correction. The primary parameters were configured as follows: minimum peak height set at 500, signal-to-noise ratio at 5, and Gaussian smooth width established as 1. The area under each peak represented the relative content of the corresponding metabolite.
Metabolome analysis
Partial least squares discriminant analysis (PLS-DA) was conducted using metaX (v.2.71)47. Univariate analysis (t-test) was applied to determine statistical significance. Metabolites with VIP > 1, P-value < 0.05, and |log2FC| ≥ 1.585 (i.e., fold change ≥ 3 or ≤ 0.333) were identified as differential metabolites. Pearson correlation analysis was performed using the WGCNA package (v.1.71) to determine the correlation between QC samples. Principal Coordinates Analysis (PCoA) was performed to reduce the dimension of the original variables using the vegan package (v.2.6-2) in R (v.4.1.3). The statistical significance of metabolite differences between poplar genotypes was assessed by PERMANOVA using the vegan package.
Establishment of the co-expression network
To explore the intricate genetic regulatory network of poplar mediating microbial community structure, the R package cluster (v.2.1.4) with the k-means method was used to analyze the co-expression/co-regulation of flavonoids in root samples of nine poplar species. Rigorous Pearson’s correlation analysis (r ≥ 0.7, P-values < 0.01) was applied to identify the DEGs and ASVs significantly associated with each flavonoid.
Data Records
The raw amplicon data are publicly accessible in the NCBI BioProject accession number PRJNA124148948. The raw root transcriptome data could be accessed under PRJNA124142449. Data products derived from the raw sequence data and raw metabolomics data are available in figshare (https://figshare.com/; https://doi.org/10.6084/m9.figshare.28775111)50. They have been appropriately specified in the text where required.
Technical Validation
Experimental treatment
To ensure the reliability of the experimental results, we cloned different poplar species at the same time and transplanted them into the natural soil at the same time to maintain the consistency of the initial growth of the plants. To reduce some objective interference, five seedlings of each species were transplanted (except Peu-H). During sample collection and phenotypic determination, one seedling with the highest and one with the lowest was removed, and the three seedlings with the comparable growth were selected as biological replicates. Due to the small root system of Peu-H, ten seedlings were transplanted, and the two highest and two lowest plants were removed, the two individual plants were pooled as one biological replicate.
Samples were taken aseptically by using sterilized equipment and sterile RNase and DNase-free tubes. Plant RNA and soil DNA were extracted in an RNase free environment. During the library preparation quantity and quality of the nucleic acids were measured with a Qubit 2.0 Fluorometer (Invitrogen, USA) and 2100 Bioanalyzer (Agilent Technologies, USA), respectively. Poplar RNA integrity was accurately detected with Fragment Analyzer 5400 (Agilent Technologies, USA), and sequencing was performed after qualified quality.
Data quality control
Since metabolites in metabolomics are easily disturbed by external factors and change rapidly, quality control during sample testing is particularly important for the accuracy of data results. The metabolite data were validated for accuracy and reliability by four QC samples. The correlation between QC samples was 0.974–0.981 (Fig. 3a), indicating that the entire detection process was stable and the data quality was high. A PCoA showed clear separation, with samples from each section combination clustered together (PERMANOVA, R2 = 0.49, P-values < 0.01; Fig. 3b).

Characterization of the metabolomic data. (a) Pearson correlation analysis between quality control (QC) samples. (b) PCoA analysis of the metabolic samples (PERMANOVA, R2 = 0.49, P-values < 0.01).
The result showed that Mean Quality Scores and Sequence Quality Scores of the transcriptome (Table 2) and amplicon sequencing (Table 3) results were of excellent quality. The GC contents were consistent at 43.70–44.90% and 53.72–56.78%, respectively, and the Q30 of the two sequencing reads reached above 93% and 94%, respectively. Moreover, the mismatch rates of sequence mapping for transcriptome analysis were between 0.07–0.28% (Table 4).
Usage Notes
To take full advantage of our dataset and further evaluate its effectiveness, we established a comprehensive gene-flavonoid-microbe co-expression network. 110 differential flavonoids (fold change ≥ 3 or ≤ 0.333, P-values < 0.05; Supplementary Data 1) were identified and further classified into six clusters based on their accumulation patterns using the k-means clustering algorithm. Next, a rigorous correction (r ≥ 0.7, P-values < 0.01) was used to screen for differentially expressed genes (DEGs; |log2FC| > 1, FDR adjusted P-values < 0.05; Supplementary Data 2) and microorganisms significantly associated with each flavonoid, and to correlate gene expression patterns, microbial enrichment patterns with flavonoid accumulation (Fig. 4a). The 110 differential flavonoids, 17,698 DEGs, and 2,579 ASVs were further classified into six co-expression clusters based on Pearson correlation analysis (Table 5 and Supplementary Data 3), and genes, flavonoids, and microorganisms in these clusters showed a unified and clear abundance pattern.

Multi-omics network about the comprehensive gene-metabolite-microbe associations. (a) Correlation network of phenylpropanoid biosynthesis pathway, flavone and flavonol biosynthesis pathway, MYB transcription factor family, bHLH transcription factor family, flavonoids, and microbes. Highly correlated associations (r ≥ 0.7, P-values < 0.01) were present. Pearson’s correlation between Cellvibrionaceae (b), Pseudomonadaceae (c), and Burkholderiaceae (d) and the height (cm) and fresh shoot biomass (g) of poplars.
Functional enrichment analyses showed that all gene clusters were enriched with flavonoid synthesis-related pathways (phenylalanine metabolism, phenylpropanoid biosynthesis, and flavonoid biosynthesis; Table 5 and Supplementary Data 4). Moreover, two genes (2/3) of flavone and flavonol biosynthesis pathway were present in Cluster II (Table 5). Correspondingly, nine (8/17) flavones and two flavonols (3/17) were observed in Cluster II (Table 5). Among them, Naringenin (methylNaringenin C-pentoside) could recruit beneficial bacteria Aeromonas sp. H1 increased plant dehydration resistance. Apigenin (Apigenin-6,8-di-C-glycoside, Apigenin C-pentoside, and methylApigenin C-hexoside) plays an important role in the recruitment of Rhizobium, Oxalobacteraceae, and Pseudomonas17,51,52. Notably, members of the MYB, bHLH, and cytochrome P450 gene families were observed in all clusters (Table 5). These gene families have been previously shown to be widely involved in the biosynthesis of flavonoids, with members MYB72 and MYC2 (bHLH) being able to shape the structure of plant-associated microbial communities11,53. We noticed that several microbes were present in the microbe cluster, such as Cluster I (Bacillaceae and Oxalobacteraceae) and Cluster IV (Pseudomonadaceae, Enterobacteriaceae, and Aeromonadaceae; Supplementary Data 3). These microbes have been reported to be associated with plant nutrient uptake and&or stress responses20,52,54,55,56. Together, these findings suggest that the dataset and co-expression network we established can provide a good basis for studying the genetic mechanisms of poplar regulation of flavonoid-mediated microbial recruitment.
We next demonstrate through subnetwork analysis how co-expression networks can be used to identify novel transcription factors that mediate rhizosphere microbial structure. In Cluster IV, a large number of flavones and flavonols are gathered, which were related to Cellvibrionaceae, Pseudomonadaceae, Burkholderiaceae, and Streptomycetaceae (Fig. 4a). Further analysis showed that Cellvibrionaceae and Pseudomonadaceae were significantly positively correlated with fresh shoot biomass and height of poplar (P-values < 0.01; Fig. 4b–d). Therefore, screening out new transcription factors that regulate the synthesis of flavones or flavonols is of great significance for achieving microbial regulation of poplar growth and development. Cluster IV contains genes such as bHLH74 and MYB4 (Supplementary Data 3), which have been shown to be involved in the anthocyanin synthesis57,58, but their roles in the synthesis of flavones and flavonols are still unclear. bHLH74 and MYB4 are significantly correlated with flavones and flavonols (r > 0.7, P-values < 0.01; Fig. 4a), suggesting that they may play an irreplaceable role in regulating the synthesis of flavones and flavonols.
Code availability
No custom code was generated for this work.
References
French, E., Kaplan, I., Iyer-Pascuzzi, A., Nakatsu, C. H. & Enders, L. Emerging strategies for precision microbiome management in diverse agroecosystems. Nat. Plants. 7, 256–267 (2021).
Liu, Y. et al. Root colonization by beneficial rhizobacteria. FEMS Microbiol. Rev. 48, 1–20 (2024).
Wang, L. et al. Multifaceted roles of flavonoids mediating plant-microbe interactions. Microbiome. 10, 233 (2022).
Wagner, M. R. Prioritizing host phenotype to understand microbiome heritability in plants. New Phytol. 232, 502–509 (2021).
Cheng, S. et al. Evolutionarily conserved core microbiota as an extended trait in nitrogen acquisition strategy of herbaceous species. New Phytol. (2024).
Fitzpatrick, C. R. et al. Assembly and ecological function of the root microbiome across angiosperm plant species. Proc. Natl. Acad. Sci. USA. 115, E1157–E1165 (2018).
Wei, M. et al. PuHox52-mediated hierarchical multilayered gene regulatory network promotes adventitious root formation in Populus ussuriensis. New Phytol. 228, 1369–1385 (2020).
Liu, Q., Cheng, L., Nian, H., Jin, J. & Lian, T. Linking plant functional genes to rhizosphere microbes: a review. Plant Biotechnol. J. 21, 902–917 (2023).
Zhang, J. et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 37, 676–684 (2019).
Liu, L. et al. Transgenic soybean of GsMYB10 shapes rhizosphere microbes to promote resistance to aluminum (Al) toxicity. J. Hazard. Mater. 455, 131621 (2023).
Stringlis, I. A. et al. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA. 115, E5213–E5222 (2018).
Singh, G., Agrawal, H. & Bednarek, P. Specialized metabolites as versatile tools in shaping plant-microbe associations. Mol. Plant. 16, 122–144 (2023).
Yang, K. et al. RIN enhances plant disease resistance via root exudate-mediated assembly of disease-suppressive rhizosphere microbiota. Mol. Plant. 16, 1379–1395 (2023).
Korenblum, E., Massalha, H. & Aharoni, A. Plant-microbe interactions in the rhizosphere via a circular metabolic economy. Plant Cell. 34, 3168–3182 (2022).
Wasson, A. P., Pellerone, F. I. & Mathesius, U. Silencing the Flavonoid Pathway in Medicago truncatula Inhibits Root Nodule Formation and Prevents Auxin Transport Regulation by Rhizobia. Plant Cell. 18, 1617–1629 (2006).
Shaw, L. J., Morris, P. & Hooker, J. E. Perception and modification of plant flavonoid signals by rhizosphere microorganisms. Environ. Microbiol. 8, 1867–1880 (2006).
Janczarek, M., Rachwał, K., Marzec, A., Grządziel, J. & Palusińska-Szysz, M. Signal molecules and cell-surface components involved in early stages of the legume-rhizobium interactions. Applied Soil Ecology: A Section of Agriculture, Ecosystems & Environment. 85, 94–113 (2015).
Liu, C. & Murray, J. D. The Role of Flavonoids in Nodulation Host-Range Specificity: An Update. Plants. 5, 33 (2016).
Yan, D. et al. Genetic modification of flavone biosynthesis in rice enhances biofilm formation of soil diazotrophic bacteria and biological nitrogen fixation. Plant Biotechnol. J. 20, 2135–2148 (2022).
He, D. et al. Flavonoid-attracted Aeromonas sp. from the Arabidopsis root microbiome enhances plant dehydration resistance. The ISME J. 16, 2633 (2022).
Zhou, C. J. et al. Evaluation of genetic diversity and germ plasm identification of 44 species, clones, and cultivars from 5 sections of the genus Populus based on amplified fragment length polymorphism analysis. Ptant Molecular Biology Reporter. 23, 39–51 (2005).
Gan, H. et al. Phosphorus and nitrogen physiology of two contrasting poplar genotypes when exposed to phosphorus and/or nitrogen starvation. Tree Physiol. 36, 22–38 (2016).
Cregger, M. A. et al. The Populus holobiont: dissecting the effects of plant niches and genotype on the microbiome. Microbiome. 6, 31 (2018).
Van Nuland, M. E. et al. Above- and belowground fungal biodiversity of Populus trees on a continental scale. Nat. Microbiol. 8, 2406–2419 (2023).
Veach, A. M. et al. Rhizosphere microbiomes diverge among Populus trichocarpa plant-host genotypes and chemotypes, but it depends on soil origin. Microbiome. 7, 76 (2019).
Moreau, D. et al. A plant perspective on nitrogen cycling in the rhizosphere. Funct. Ecol. 33, 540–552 (2019).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 9, 671–675 (2012).
Qingxue Guo, L. Y. H. K. Plant-plant interactions and N fertilization shape soil bacterial and fungal communities. Soil Biol. and Biochem. 128, 127–138 (2019).
Geng, J. et al. Determination of the Best Controlled-Release Potassium Chloride and Fulvic Acid Rates for an Optimum Cotton Yield and Soil Available Potassium. Front. Plant Sci. 11, 562335 (2020).
McCormack, M. L. et al. Redefining fine roots improves understanding of below-ground contributions to terrestrial biosphere processes. New Phytol, 505–518 (2015).
Bulgarelli, D. et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 488, 91–95 (2012).
Idbella, M. et al. Negative plant-soil feedback in Arabidopsis thaliana: Disentangling the effects of soil chemistry, microbiome, and extracellular self-DNA. Microbiol. Res. 281, 127634 (2024).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7, 335–336 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 26, 2460–2461 (2010).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Edgar, R. C. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. Biorxiv, 81257 (2016).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic. Acids. Res. 41, D590–D596 (2013).
Lima, J. et al. Taxonomic annotation of 16S rRNA sequences of pig intestinal samples using MG-RAST and QIIME2 generated different microbiota compositions. J. Microbiol. Methods. 186, 106235 (2021).
Lu, J. & Salzberg, S. L. Ultrafast and accurate 16S rRNA microbial community analysis using Kraken 2. Microbiome. 8, 124 (2020).
Li, Y. et al. MicroTom Metabolic Network: Rewiring Tomato Metabolic Regulatory Network throughout the Growth Cycle. Mol. Plant. 13, 1203–1218 (2020).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, i884–i890 (2018).
Tuskan, G. A. et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science. 313, 1596–1604 (2006).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32, 292–294 (2016).
Akimoto, N. et al. FlavonoidSearch: A system for comprehensive flavonoid annotation by mass spectrometry. Sci. Rep. 7 (2017).
Wen, B., Mei, Z., Zeng, C. & Liu, S. metaX: a flexible and comprehensive software for processing metabolomics data. BMC Bioinformatics. 18, 183 (2017).
NCBI Sequence Read Archive https://identifers.org/ncbi/insdc.sra:SRP572791 (2025).
NCBI Sequence Read Archive https://identifers.org/ncbi/insdc.sra:SRP572846 (2025).
Wu, J., Zhang, D. & Xie, J. An integrated transcriptome, metabolome, and microbiome dataset of Populus under nutrient-poor conditions. Figshare https://doi.org/10.6084/m9.figshare.28775111.v1 (2025).
Yu, X. Q., Yan, X., Zhang, M. Y., Zhang, L. Q. & He, Y. X. Flavonoids repress the production of antifungal 2,4-DAPG but potentially facilitate root colonization of the rhizobacterium Pseudomonas fluorescens. Environ. Microbiol. 22, 5073–5089 (2020).
Yu, P. et al. Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nat. Plants. 7, 481–499 (2021).
Hou, S. et al. A microbiota-root-shoot circuit favours Arabidopsis growth over defence under suboptimal light. Nat. Plants. 7, 1078–1092 (2021).
Li, Q. et al. Plant growth-promoting rhizobacterium Pseudomonas sp. CM11 specifically induces lateral roots. New Phytol. 235, 1575–1588 (2022).
Li, Y. et al. Volatile compounds from beneficial rhizobacteria Bacillus spp. promote periodic lateral root development in Arabidopsis. Plant Cell Environ. 44, 1663–1678 (2021).
Madhaiyan, M. et al. Improvement of plant growth and seed yield in Jatropha curcas by a novel nitrogen-fixing root associated Enterobacter species. Biotechnol. Biofuels. 6, 140 (2013).
Wang, X. C. et al. Arabidopsis MYB4 plays dual roles in flavonoid biosynthesis. Plant J. 101, 637–652 (2020).
Song, Y. et al. UV-B induces the expression of flavonoid biosynthetic pathways in blueberry (Vaccinium corymbosum) calli. Front. Plant Sci. 13, 1079087 (2022).
Acknowledgements
We are particularly grateful to Professors Francis Martin, Ton Bisseling, and Huchen Li for their careful guidance, insightful discussions, and constructive ideas on this study. This work was supported by funding from the Fundamental Research Funds for the National Key R&D Program of China (nos. 2022YFD2201600, 2022YFD2200602, and 2023YFD2200203), the Project of the National Natural Science Foundation of China (nos. 32371906 and 32022057), Forestry and Grassland Science and Technology Innovation Youth Top Talent Project of China (no. 2020132607), Central Universities [QNTD202305, BFUKF202413], the 111 Project [No.B20050].
Author information
Authors and Affiliations
Contributions
J.X. and D.Z. designed the experiments. J.W., D.H., S.L., S.T. and H.W. performed the experiments. J.W., Y.W., D.H. and S.L. collected and analyzed the data. J.W. and J.X. wrote the manuscript. J.W., Y.W., D.H., S.L., S.T., Y.D. and H.W. revised the manuscript. J.X. and D.Z. obtained funding and are responsible for this article. All authors read and approved the manuscript. J.W., D.H. and Y.W. contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, J., He, D., Wang, Y. et al. An integrated transcriptome, metabolome, and microbiome dataset of Populus under nutrient-poor conditions. Sci Data 12, 717 (2025). https://doi.org/10.1038/s41597-025-05029-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05029-1
