
Abstract
Parotis Hübner, 1831 is a genus within the family Crambidae, which is recognized as one of the most diverse families of Lepidoptera. Species within the genus Parotis can be readily distinguished from other closely related genera by their distinctive green or yellow-green body coloration. However, the genus Parotis has received relatively limited research attention, and the scarcity of genome-wide molecular resources has impeded a more comprehensive understanding of its evolution, adaptation, and phylogenetic relationships. This study reports the first genome assembly for Parotis chlorochroalis (Hampson, 1912), generated through PacBio Hi-Fi and Hi-C sequencing technologies. The assembled genome has a size of 456.23 Mb, comprising 31 chromosomes. Approximately 181.82 Mb, which constitutes 39.85% of the genome, has been identified as repetitive sequences. The genome assembly includes 16,299 protein-coding genes, of which 94.82% have been functionally annotated. This chromosome-level genome assembly not only advance understanding of P. chlorochroalis but also has the potential to facilitate genomic studies of other lepidopteran species.
Similar content being viewed by others


Background & Summary
Crambidae is one of the most speciose families of Lepidoptera, currently containing 15 subfamilies, 1,015 genera and over 11,500 species globally1,2,3. Many species are economically important pests, affecting crops and stored food products. Spilomelinae is one of the species-rich subfamilies with 4,135 described species belonging to 344 genera worldwide, it is the most speciose group among pyraloids3. Their host plants range from ferns4 over gymnosperms5 to a wide spectrum of angiosperms. Many Spilomelinae tribes have a narrow food spectrum, with the larvae feeding on plants of only one or a few plant families6, including a variety of economically important crops.
Species of the genus Parotis Hübner, 1831 from the subfamily Spilomelinae are easily distinguishable taxa with typical morphological characters that uniform the whole body with green or yellow-green color. This genus comprises 43 recognized species distributed across the Palaearctic, Oriental, and Australian regions, with 15 species documented in China, particularly in the southern region. Parotis larvae are leaf-folders, which fold both sides of a leaf to be a bag-like shape, host plants including Rubiaceae, Apocynaceae and Euphorbiaceae7,8. Larvae of Parotis prefer to feed on tender leaves as a window-feeder by removing discrete patches of mesophyll and overlying epidermis to avoid the latex secreted veins7. Despite its taxonomic distinctiveness, Parotis has received relatively limited research attention, with most studies focusing on species identification and taxonomic revisions. The limited genomic resource extremely hinders deeper understanding of evolution, adaptation, and phylogenetic relationships of this genus.
Parotis chlorochroalis was first described by Hampson (1912)9, and distributes in Cameroon, Nigeria Congo10,11,12 and China13. This species is characterized by its pale green body, fulvous-marked palpi, and slight fulvous stripes on the shoulders. The forewings have a pale fulvous costal edge with black discoidal and terminal points, while the hindwings also feature a black discoidal point, both with whitish cilia. Males possess a prominent fuscous-black anal tuft mixed with silvery scales. To enhance the understanding of the evolution and ecology of Parotis, a chromosome-level genome of P. chlorochroalis (Hampson, 1912) was obtained through the combination of PacBio Hi-Fi long reads, Illumina short reads, and Hi-C data. The repeats, non-coding RNAs (ncRNAs), and protein-coding genes (PCGs) were annotated, and conducted gene family evolution analysis. The high-quality genome of P. chlorochroalis is an important milestone in understanding of Parotis and will contribute to the study of Parotis evolution and ecology.
Methods
Sample collection and sequencing
The P. chlorochroalis samples used in this study were collected in Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences in Yunnan Province, China on 15 July 2022 (Figure S1). Adult individuals were collected by light-trap, brought back to laboratory alive, and stored in −80 °C freeze after instantly freezing with liquid nitrogen. Two male adult specimens were used for PacBio Hi-Fi and Hi-C sequencing, one female specimen for transcriptome sequencing. Besides, to identify the sexual link chromosome, both one male and female adult specimen were used for genome survey sequencing. Genomic DNA and RNA from specimens were extracted using the DNeasy Blood & Tissue Kit and TRIzoTM Reagent, following the manufacturer’s instructions. The abdomen of all specimens was removed before DNA extraction to avoid contamination of intestinal contents. PCR-free short-read libraries of 150 bp paired-end read with a 350 bp insert size were generated using the Truseq DNA PCR-free Kit. The Hi-C sequencing was carried out by digesting extracted DNA with the Mbol restriction enzyme. The Illumina NovaSeq6,000 platform was utilized to sequence all short-read libraries.
After examination of the quality of isolated DNA, the library of 15 kb was constructed using a SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, CA, USA). The construction included DNA shearing, AMPure PB Bead purification, ssDNA overhangs removing, damage repair, end repair, hairpin adapter ligation, and bead purification of the library. After quality control test, a SMRTbell library was obtained. The library was sequenced using a single 8 M SMAT Cell on the PacBio Sequel II platform (Pacific Biosciences, CA, USA) (PacBio Sequel II System).
Berry Genomics (Beijing, China) carried out all library construction and sequencing. Finally, a total of 384.50 Gb of sequencing data were obtained, comprising 37.63 Gb (82.48 × coverage) of PacBio Hi-Fi reads, 71.48 GB of Illumina reads (31.72 GB (69.53×) for male, 39.76 GB (87.15x) for female), 54.03 Gb (118.43 × coverage) of Hi-C data, and 6.89 Gb of transcriptome data (Table 1). The raw PacBio Hi-Fi reads had a scaffold N50 and an average length of 17.53 and 17.74 kb, respectively.
Genome size estimation and assembly
Quality control on raw Illumina data performed using fastp v0.23.214 using default parameters. The strategy of short-read k-mer distributions was employed to estimate the genome size. The histogram of k-mer frequencies was computed with 17-mers using Jellyfish v2.3.015, and the k-mer histogram was provided to the R package findGSE v1.016 to estimate the genome size. As a result, the genome size was estimated to be 460.17 Mb (Figure S2).
The primary assembly of PacBio Hi-Fi long reads was generated using hifiasm v 0.16.1-r37517 and wtdbg2 v2.518. The haplotypic duplication was identified and removed with purge_dups v1.2.51319 for hifiasm assembly. NextPolish v1.4.120 was used to polish the wtdbg2 assembly with Illumina and PacBio reads. After then, two assemblies were merged using quickmerge v0.321. Hi-C reads were aligned to the merged assembly after performing quality control using Juicer v1.622. Subsequently, contigs were anchored onto chromosomes using 3D-DNA v180923. To ensure accuracy, manually review and correction were performed with Juicebox v1.11.0824.
Potential contaminants were screened using blastn (BLAST + v2.11)25 against the NCBI nucleotide database, and sequences shorter than 1,000 bp and non-target sequences were filtered using BlobToolKit environment26. Besides, univec contaminants regions were removed by alignment against the UniVec database27, and the final non-contaminated genome assembly was extracted using seqkit v2.2.028 and bedtools v2.30.029.
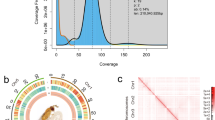
The final chromosome-level genome assembly of P. chlorochroalis had a size of 456.23 Mb, comprising 362 scaffolds and 569 contigs, with the scaffold and contig N50 sizes of 16.46 Mb and 15.53 Mb, respectively. Among them, 178 contigs (98.62%, 449.84 Mb) were anchored into 31 chromosomes with lengths ranging from 7.16 to 69.80 Mb and the GC content was 37.08% (Table 2, 3; Figs. 1, 2). The genome completeness was assessed with BUSCO v 5.4.230 with the reference lepidoptera gene set (n = 5,286). The final genome assembly showed a BUSCO completeness of 96.1%, consisting of 946 (93.4%) single-copy BUSCOs, 2.7% were duplicated, 0.7% were fragmented, and 3.2% were missing. The mapping rates of PacBio, Illumina, and RNA reads to the genome were 99.90%, 98.52% (female) /97.89% (male), and 97.71%, respectively.

Genome-wide chromosomal heatmap of Parotis chlorochroalis, with each chromosome and contig framed in blue and green, respectively.

Genome characteristics of Parotis chlorochroalis. From the outer ring to the inner ring are the distributions of chromosome length, gene density, GC content, simple repeats and TEs (LINE, SINE, DNA, and LTR).
Sex chromosomes detection
To identify sex-linked fragments in the genome, high-quality clean reads were mapped to the pseudochromosome sequences using bwa v0.7.1731 and samtools v1.15.132. The depth coverages of male and female samples were calculated using bamdst v1.0.9 (https://github.com/shiquan/bamdst). Subsequently, the male to female (M: F) coverage ratio calibrated by the average depth coverages was used to determine sex-linked scaffolds. Chromosome 1 with a log2 (F: M coverage ratio) value approximately 1 was defined as Z chromosome (Z chromosome possess approximately twice greater coverage in male than in female), other pseudochromosome with value approximately 0 as autosome (Figure S3).
Genome annotation
A custom repeat library was generated using RepeatModeler v2.0.333. RepeatMasker v4.1.2-p134 was utilized to identify repetitive elements in the P. chlorochroalis genome by aligning it against the custom library. The analysis revealed that the P. chlorochroalis genome contains approximately 39.85% (181.82 Mb) repetitive elements, comprising unknow elements (8.61%), LTR elements (1.21%), DNA transposons (2.26%), LINE (11.48%), SINE (5.00%), simple repeats (0.87%) (Table 3), as well as other elements (Table S1). Furthermore, sequence divergence estimates revealed a peak at low divergence rates (∼1%) in TE sequences of P. chlorochroalis, indicating a recent expansion of TEs (Figure S4).
Non-coding RNAs (ncRNAs) and tRNAs were identified using Infernal v1.1.435 and tRNAscan-SE v2.0.936, respectively. The low-confidence tRNAs were filtered using EukHighConfidenceFilter from tRNAscan-SE. A total of 817 ncRNAs in the genome of P. chlorochroalis were identified (Table 3), including 90 ribosomal RNAs, 81 microRNAs, 101 small nuclear RNAs, 478 transfer RNAs, four ribozymes, and 63 other ncRNAs (Table S2).
Protein-coding genes (PCGs) were annotated using MAKER v3.01.0337 based on three strategies, containing ab initio predictions, homology-based, and transcriptome-based approaches. To maximize ab initio predictions, the BRAKER v2.1.638 were employed with transcriptome and protein evidence, and combined their results as the ab initio input for MAKER. BRAKER used Augustus v3.4.039 and GeneMark-ES Suite 4.71_lic40 as predictors and automatically trained them from reference proteins mined from OrthoDB v10 database41. Protein sequences from five species (Apis mellifera (GCF_003254395.2), Drosophila melanogaster (GCF_000001215.4), Bombyx mori (GCF_030269925.1), Chilo suppressalis (GCA_902850365.2), and Diatraea saccharalis (GCA_918026875.4)) were used for homologous gene annotation. The transcriptome used for MAKER pipeline was assembled under a genome-guided method via HISAT2 v2.1.142 and StringTie v2.2.143, and redundant isoforms were removed with cdhit v4.8.144.
The final annotation predicted 16,299 protein coding genes, with an average length of 7291.36 bp for genes (Table 3). The average number of exons, introns, and CDS of each gene were 6.44, 5.44, and 6.33, respectively, and their corresponding mean length was 301.59, 983.57, and 223.71 bp, respectively (Table S3). BUSCO completeness of the protein sequences was 91.6% (n = 5,286), including 90.7% (4797) single-copy, 0.9% (47) duplicated, 1.9% (101) fragmented, and 6.5% (341) missing BUSCOs, indicating high-quality predictions.
Gene functional annotation was conducted by searching against the UniProtKB database45 using Diamond v2.0.15.15346 in sensitive mode with the parameters “--sensitive -e 1e-5”. eggNOGmapper v2.1.1247 and InterProScan 5.53–87.048 were employed to assign Gene Ontology (GO) and (KEGG, Reactome) pathway annotations and to identify protein domains. The InterProScan analyses included five databases: Pfam49, SMART50, Superfamily51, Gene3D52, and CDD53. The results predicted by the above tools were integrated to obtain the final gene function prediction. Genes with 8,141 GO terms, 4,176 KEGG pathways, 3,032 Reactome pathways, 2,430 Enzyme Codes, and 11,690 COG categories were assigned by integrating the InterProScan and eggNOG annotation results (Table S3).
Data Records
The raw sequencing data and genome assembly of P. chlorochroalis have been deposited at the Genome Sequence Archive54 and Genome Warehouse55 in National Genomics Data Center (NGDC)56. The raw sequence data of Illumina, transcriptome, Hi-C, and PacBio can be found under identification numbers CRR1371036-CRR137104057,58,59,60,61 for NGDC and SRP50644662 for NCBI, the whole genome sequence assembly can be found under identification numbers GWHFIDL00000000.163 as well as deposited in the NCBI assembly with the accession number GCA_047302205.164. Additionally, the results of annotation for repeated sequences, gene structure, functional prediction and supplementary files have been deposited in the ScienceDB database65.
Technical Validation
Phylogeny and gene family evolution
Orthology analyses was performed on PCG sequences across 26 Lepidoptera species, comprising 21 moth species and five butterfly species (Table 4). The redundant isoforms were eliminated using cdhit (-c 0.98), after then orthogroup (gene families) inference using OrthoFinder v2.5.566 with Diamond mode (“-S diamond”) for sequence alignment. A total of 417,889 (93.53%) genes were assigned to 20,282 orthogroups, of which 3,231 were shared by all eight species and 454 were single-copy genes (Table S4). For P. chlorochroalis, 15,190 genes (93.20%) were contained in 10,600 gene families, of which 49 families and 808 genes were specific to this species.
Single-copy orthologues identified by OrthoFinder were aligned using MAFFT v7.49067 with the high-accuracy LINS-I strategy. Alignment gaps were removed using trimAl v1.4.168 with the “automated1” parameter, and all sequences were concatenated using FASconCAT-G v1.0469. Finally, the phylogenetic tree was reconstructed on the single-copy orthologs using IQ-TREE v2.0770 with the LG site-homogeneous model. The ultrametric tree was transformed using r8s v1.8.171 and the time-calibrated by the divergence time between Cnaphalocrocis medinalis and Chilo suppressalis (68.8 Mya), Danaus plexippus and Kallima inachus (72.9 Mya), Bombyx mori and Manduca sexta (74.5 Mya), Pieris napi and Zerene cesonia (69.5 Mya) from the TimeTree database72. The analysis showed that P. chlorochroalis is closely related to Cnaphalocrocis medinalis, which both belong to subfamily Spilomelinae, and six Crambidae species format a cluster (Fig. 3). The phylogenetic results were consistent with previous studies, supporting Pyralidae (Ephestia elytella) as a sister group to Crambidae73,74 (Fig. 3).

The phylogeny and gene family changes among 26 Lepidoptera species. The divergence times were estimated by r8s using the calibration time from Timetree. The values labeled at terminals denote the number of significantly expanded and contracted gene families. “1:1:1” represents universal single-copy genes in all species, “N:N:N” represents multi-copy genes, “others” represents unclassified orthologues, and “unassigned” represents orthologues that cannot be assigned to any orthogroups.
Gene family evolution (expansion or contraction) was estimated using CAFÉ v4.2.175 based on the generated phylogenetic tree, revealing 820 expanded and 1,449 contracted gene families in P. chlorochroalis, including 39 gene families that underwent rapid evolution (35 expansions and 4 contractions). The significantly expanded families included the Reverse transcriptase, CCHC-type ___domain-containing protein, Ribonuclease H protein, Chitin-binding and other families that play important roles in the development, metabolism, and adaptive evolution of P. chlorochroalis (Table S5). Subsequently, functional enrichment (GO and KEGG) analysis on PCGs from significantly expanded families were performed using ClusterProfiler v4.0.176 with default parameters. The enrichment of GO and KEGG in rapidly expanding families further indicates their function in the membrane biogenesis, cell-cell junction, lipid biosynthesis, and signaling pathway, among others (Figure S5a,b).
Chromosome synteny
To investigate interspecific chromosomal evolution, the genome of P. chlorochroalis was compared against with that of B. mori and D. saccharalis. The pairwise synteny were searched, filtered, and visualized using JCVI77, the subset of blocks were extracted with following options “--minspan = 30 --simple”. Syntenic analyses showed that 80 syntenic blocks (8,500 gene pairs contained 16,998 collinear genes) between P. chlorochroalis and B. mori and 99 syntenic blocks (8,851 gene pairs contained 17,694 collinear genes) between P. chlorochroalis and D. saccharalis were conserved. The average number of genes per block was 106 and 89, while a notable 33.75% (27 blocks) and 23.23% (23 blocks) contained over 100 collinear genes for P. chlorochroalis vs. B. mori and P. chlorochroalis vs. D. saccharalis, respectively. Notably, the analysis revealed that the three B. mori chromosomes 11, 23 and 24 were clearly divided into three pairs in P. chlorochroalis: 10 and 29, 13 and 30, 27 and 31, respectively (Fig. 4). The D. saccharalis chromosomes 1~5, 7, 8 and 20 were divided into eight pairs in P. chlorochroalis: 1 and 21, 16 and 18, 17 and 22, 8 and 24, 20 and 25, 7 and 23, 19 and 26, 27 and 31, respectively; D. saccharalis chromosomes 6 divided into 3 chromosomes in P. chlorochroalis (13, 28 and 29); while D. saccharalis chromosomes 22 and 23 merged into one chromosome in P. chlorochroalis (30); D. saccharalis chromosomes 21 should be the chromosome W which not represented in current P. chlorochroalis genome assembly.

Chromosomal synteny between Parotis chlorochroalis, Diatraea saccharalis and Bombyx mori.
Code availability
All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software. The main script is available in the ScienceDB database65.
References
Léger, T., Mally, R., Neinhuis, C. & Nuss, M. Refining the phylogeny of Crambidae with complete sampling of subfamilies (Lepidoptera, Pyraloidea). Zoologica Scripta 50, 84–99 (2021).
Powell, J. A. in Encyclopedia of Insects (Second Edition) (eds Vincent, H. R. & Ring, T. C.). pp559–587 (Academic Press, 2009).
Nuss, M. et al. Global Information System on Pyraloidea., www.pyraloidea.org (2003–2024).
Farahpour-Haghani, A., Jalaeian, M. & Landry, B. Diasemiopsis ramburialis (Duponchel) (Lepidoptera, Pyralidaes. l., Spilomelinae) in Iran: first record for the country and first host plant report on water fern (Azolla filiculoides Lam., Azollaceae). Nota lepidopterologica 39, 1–11 (2016).
Inoue, H. & Yamanaka, H. Redescription of Conogethes punctiferalis (Guenée) and descriptions of two new closely allied species from Eastern Palaearctic and Oriental Regions (Pyralidae, Pyraustinae). Tinea 19, 80–91 (2006).
Mally, R., Hayden, J., Neinhuis, C., Jordal, B. & Nuss, M. The phylogenetic systematics of Spilomelinae and Pyraustinae (Lepidoptera: Pyraloidea: Crambidae) inferred from DNA and morphology. Arthropod Systematics & Phylogeny 77, 141–204 (2019).
Lin, C. S. Parotis Hübner (Lepidoptera: Crambidae) of Taiwan. Journal of Taiwan Museum 50, 33–46 (1997).
Common, I. F. B. Moths of Australia. (CSIRO Publishing, 1990).
Hampson, G. F. Descriptions of new species of Pyralidae of the subfamily Pyraustinae. Vol. 10 1–20, 557–573 (1912).
Meyrick, E. Exotic Microlepidoptera Taylor and Francis, London, 1–642 (1930-1936).
J, G. Lépidoptères Microlépidoptères (deuxième partie). [Annales du Musée du Congo Belge, Zoologie [3, Arthropodes] Section 2. Catalogues Raisonnés 7, 121–240 (1942).
T, J. A. J. List of species of Pyralidae. Collected by Alexander Barns T., Central Africa, 1919, 1920, 1921. Bulletin of the Hill Museum: A Magazine of Lepidopterology 1, 486 (1924).
Yang, Y. A taxonomic study on the genera of Parotis Hübner,1831 and Conogethes Meyrick,1884 of China (Lepidoptera: Crambidae: Spilomelinae), (2021).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Sun, H., Ding, J., Piednoël, M. & Schneeberger, K. findGSE: estimating genome size variation within human and Arabidopsis using k-mer frequencies. Bioinformatics 34, 550–557 (2018).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat Methods 17, 155–158 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Chakraborty, M., Baldwin-Brown, J. G., Long, A. D. & Emerson, J. J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res 44, e147–e147, https://doi.org/10.1093/nar/gkw654 (2016).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell systems 3, 99–101 (2016).
Ye, J., McGinnis, S. & Madden, T. L. BLAST: improvements for better sequence analysis. Nucleic Acids Res 34, W6–W9 (2006).
Challis, R., Richards, E., Rajan, J., Cochrane, G. & Blaxter, M. BlobToolKit–interactive quality assessment of genome assemblies. G3: Genes, Genomes, Genetics 10, 1361–1374 (2020).
Kitts, P., Madden, T., Sicotte, H., Black, L. & Ostell, J. UniVec database. Available from: ncbi.nlm.nih.gov/VecScreen/UniVec.html (2011).
Shen, W., Sipos, B. & Zhao, L. SeqKit2: A Swiss army knife for sequence and alignment processing. iMeta, e191 (2024).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol 38, 4647–4654 (2021).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 (2013).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10 https://doi.org/10.1093/gigascience/giab008 (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. http://www.repeatmasker.org (2013–2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res 49, 9077–9096 (2021).
Campbell, M. S., Holt, C., Moore, B. & Yandell, M. Genome annotation and curation using MAKER and MAKER‐P. Current protocols in bioinformatics 48, 4.11. 11–14.11. 39 (2014).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR genomics and bioinformatics 3, lqaa108 (2021).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34, W435–W439 (2006).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR genomics and bioinformatics 2, lqaa026 (2020).
Kriventseva, E. V. et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res 47, D807–D811 (2019).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
The UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506–D515 (2018).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 18, 366–368 (2021).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol 38, 5825–5829 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res 49, D412–D419 (2021).
Letunic, I., Khedkar, S. & Bork, P. SMART: recent updates, new developments and status in 2020. Nucleic Acids Res 49, D458–D460, https://doi.org/10.1093/nar/gkaa937 (2020).
Pandurangan, A. P., Stahlhacke, J., Oates, M. E., Smithers, B. & Gough, J. The SUPERFAMILY 2.0 database: a significant proteome update and a new webserver. Nucleic Acids Res 47, D490–D494, https://doi.org/10.1093/nar/gky1130 (2018).
Lewis, T. E. et al. Gene3D: Extensive prediction of globular domains in proteins. Nucleic Acids Res 46, D435–D439 (2018).
Marchler-Bauer, A. et al. CDD: NCBI’s conserved ___domain database. Nucleic Acids Res 43, D222–D226 (2015).
Chen, T. T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics, Proteomics & Bioinformatics 19, 578–583 (2021).
Chen, M. et al. Genome Warehouse: A Public Repository Housing Genome-scale Data. Genomics Proteomics Bioinformatics 19, 584–589 (2021).
CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2024. Nucleic Acids Res 52, D18–D32 (2024).
National Genomics Data Center Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA020350/CRR1371036 (2024).
National Genomics Data Center Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA020350/CRR1371037 (2024).
National Genomics Data Center Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA020350/CRR1371038 (2024).
National Genomics Data Center Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA020350/CRR1371039 (2024).
National Genomics Data Center Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA020350/CRR1371040 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP506446 (2025).
National Genomics Data Center Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/88007/show (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_047302205.1 (2025).
Zhou, Q. S. The first chromosome-level genome assembly of Parotis chlorochroalis (Hampson, 1912) (Lepidoptera: Crambidae: Spilomelinae). Science Data Bank. https://doi.org/10.57760/sciencedb.17310 (2024).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biology 20 https://doi.org/10.1186/s13059-019-1832-y (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772–780 (2013).
Capella Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Kück, P. & Longo, G. C. FASconCAT-G: extensive functions for multiple sequence alignment preparations concerning phylogenetic studies. Frontiers in Zoology 11, 1–8 (2014).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37, 1530–1534 (2020).
Sanderson, M. J. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301–302 (2003).
Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol Biol Evol 34, 1812–1819 (2017).
Law, S. T. et al. Chromosomal-level reference genome of the moth Heortia vitessoides (Lepidoptera: Crambidae), a major pest of agarwood-producing trees. Genomics 114, 110440 (2022).
Xu, H. et al. Chromosome‐level genome assembly of an agricultural pest, the rice leaffolder Cnaphalocrocis exigua (Crambidae, Lepidoptera). Molecular Ecology Resources 22, 307–318 (2022).
Han, M. V., Thomas, G. W., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol 30, 1987–1997 (2013).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The innovation 2, 100141 (2021).
Tang, H. et al. JCVI: A versatile toolkit for comparative genomics analysis. iMeta 3, e211 (2024).
Acknowledgements
This research was supported by the National Science Foundation of China (32330013 and 32470473), the Survey of Wildlife Resources in Key Areas of Xizang (Phase II) (ZL202303601) and Sino BON Insect Diversity Monitoring Network (Sino BON-Insect).
Author information
Authors and Affiliations
Contributions
C.D.Z. and Q.S.Z. contributed to the research design. M.X. and Q.S.Z. collected the samples. M.X. and C.S.W. identified the species. M.X., B.H. and Q.S.Z. performed the genome assembly and annotation analyses. M.X., A.R.L., R.C. and B.H. analyzed the data. M.X., A.R.L., R.C., B.H. and Q.S.Z. wrote the draft manuscript and revised the manuscript. All co-authors contributed to this manuscript and approved it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, M., Cheng, R., He, B. et al. Chromosome-level genome assembly of Parotis chlorochroalis (Lepidoptera: Crambidae: Spilomelinae). Sci Data 12, 743 (2025). https://doi.org/10.1038/s41597-025-05053-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05053-1
