
Abstract
Paris polyphylla var. yunnanensis is a widely cultivated rhizomatous perennial woody plant known for producing highly valued steroidal saponins. Steroidal saponins serve as the primary active components and exhibit a variety of pharmacological effects. Their production is closely correlated with the growth environment and developmental stages of P. polyphylla. However, the medicinal value of P. polyphylla cultivated for varying durations remains unclear. In this study, we collected samples of P. polyphylla aged 3 to 8 years for metabolome and transcriptome sequencing analysis. The total saponin content was found to be significantly higher in 8-year-old P. polyphylla compared to younger plants. A total of 1,510 metabolites and 270.65 Gb of clean data were identified from these samples. This study provides insights into the medicinal values of P. polyphylla at different growth stages and elucidates the metabolic pathways for specific steroidal saponins.
Similar content being viewed by others


Background & Summary
P. polyphylla var. yunnanensis, a perennial herb of the genus Paris, is a characteristic medicinal plant of Yunnan. It is mainly distributed in Yunnan, Guizhou, and Sichuan Province. The primary chemical constituents of P. polyphylla include steroidal saponins, flavonoids, triterpenes, and polysaccharides. Notably, steroidal saponins are the most abundant chemical constituents and serve as the principal active ingredients1, and a total of 112 steroidal saponins have been isolated and identified from this species2. Research indicates that P. polyphylla possesses several pharmacological effects, including heat-clearing and toxin-detoxifying properties, anti-tumor, anti-inflammatory, antibacterial, hemostatic effects, vascular endothelial cell protection, and promotion of uterine contractions3,4. It serves as the primary raw material for over 100 proprietary Chinese medicines, including Yunnan Baiyao, Gongxuening Capsule, and Jidesheng Snake Tablet5,6. It is widely utilized in clinical settings for the treatment of hemorrhage, trauma, and chronic bronchitis2,3,6. With the rapid advancement of the Chinese herbal medicine industry, the demand for P. polyphylla as a raw material has surged significantly. Reports indicate that approximately 80% of wild resources of P. polyphylla are exploited annually, with the demand in Yunnan Province alone exceeding 1,000 tons, while the annual production of wild resources is less than 100 tons. This, coupled with the plant’s slow growth rate, results in a significant discrepancy between supply and demand. The irreversible loss of wild resources of P. polyphylla could lead to significant ecological imbalance, adversely affecting the habitat of associated flora and fauna, as well as diminishing the genetic diversity of its wild populations. Therefore, studying the growth characteristics, accumulation patterns of medicinal components, and their relationship with growth years of P. polyphylla is of great significance for the rational development and sustainable utilization of its resources7,8.
The accumulation of secondary metabolites in P. polyphylla is influenced by numerous factors. Different growing environments exhibit variations in altitude, soil composition, light exposure, and temperature, all of which significantly influence the yield and quality of P. polyphylla9. Current research has demonstrated that the number of growth years significantly influences the profiles of secondary metabolites of plants10,11,12. Currently, existing literature has conducted comparative studies on P. polyphylla from various producing areas13,14,15, different harvest periods16,17, tissue parts18,19,20 and different varieties14,15. There is also a strong correlation between saponin content and the growth stage of Paris21. However, these studies predominantly focus on single-omics approaches and lack comprehensive multi-omics integrated analyses. Transcriptome sequencing technology enables the rapid and comprehensive identification of gene expression differences and molecular marker screening in tissues or cells under specific conditions. Meanwhile, metabolomics allows for the quantitative analysis of metabolite variations within organisms, thus reflecting the outcomes of gene expression22,23,24. The integrated analysis of transcriptome and metabolomics can screen key gene expressions involved in the synthesis of secondary metabolites in medicinal plants, thereby providing a foundation for understanding their biosynthetic pathways25. Additionally, investigating the relationships between polyphyllins levels and growth years of plants will provide valuable guidance for its sustainable harvesting practices.
In this study, we collected 18 samples of P. polyphylla, aged 3 to 8 years, across six different growth stages for saponin content, transcriptome and metabolomics analyses. There was no significant difference in the total saponin content among 3, 4, 5-year-old samples, all of which had saponin levels below 0.6% (Table 1), failing to meet the standards set by the 2025 edition of the Chinese Pharmacopoeia. The highest levels of total saponins, as well as Polyphyllin I, II, and VII, were observed in the 8-year-old samples, followed by the 7-year-old samples (Table 1). A total of 270.65 Gb of clean data were obtained, and 1,510 metabolites were detected, including phenolic acids, amino acids and their derivatives, flavonoids, steroids, and so on. Among these, phenolic acids were found to be the most abundant, with 186 identified varieties, while tannins were the least abundant, with only 4 varieties detected. The contents of various compounds in P. polyphylla exhibited significant differences across different growth years (Table 2). Diosgenin serves as a precursor in the synthesis of steroidal saponins, and the peak area of diosgenin varies in P. polyphylla across different growth years. This variation may contribute to the differences in the levels of steroidal saponins. In addition, we found that the upstream genes DXS, DXR, GPPS, SMO, DWF1, CPI1, STE1, and C5-SD, of cholesterol synthesis showed significantly high expression in high-level saponin P. polyphylla (PP8). These data provide a reference for the standardized cultivation, quality control, resource protection, and sustainable utilization of P. polyphylla, which is crucial for advancing the modernization and internationalization of traditional Chinese medicine. This study offers comprehensive transcriptomic and metabolomic data for P. polyphylla at various growth stages and served as essential resources for identifying key genes participated in the saponin synthesis in P. polyphylla and for investigating their regulatory roles and functions during growth and development.
Methods
Samples collection and treatment
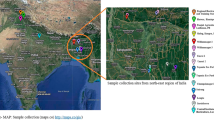
Samples of Paris polyphylla var. yunnanensis, aged between 3 to 8 years, were collected in November 2023 from Wenshan City (23.27198 N, 104.01723 E), Yunnan Province, China (Fig. 1). All selected samples were subjected to identical cultivation and management conditions. Specifically, they were planted in an open sandy loam soil field and shaded with a black plastic net that blocks 60% of the sunlight. Each sample underwent three biological replicates, and all samples were obtained from three individual plants of similar size. The samples are designated as PP3, PP4, PP5, PP6, PP7, and PP8, representing the rhizomes of P. polyphylla grown for 3, 4, 5, 6, 7, and 8 years, respectively. After removing the fibrous roots, the fresh rhizomes were washed immediately under tap water and stored at −80 °C for metabolome and transcriptome sequencing analysis.

Demonstration map of samples collection base.
Widely targeted metabolic profiling analysis
Metabolites are the basis of biological phenotype, which can help to understand biological processes and their mechanisms more intuitively and effectively. The rhizome samples were placed in a lyophilizer (Scientz-100F) utilizing vacuum freeze-drying technology. Subsequently, the samples were ground into a powder at 30 Hz for 1.5 minutes using a grinder (MM 400, Retsch). A total of 50 mg of the sample powder was extracted with 1.2 mL of 70% aqueous methanol, which had been pre-cooled to −20°C. The extraction process involved vortexing the mixture for 30 seconds every 30 minutes, repeated six times. After centrifugation at 12 000 rpm for 3 min, the supernatant was filtered through a microporous membrane (0.22 μm pore size) and stored in an injection bottle for analysis using the UPLC-ESI-MS/MS system and tandem mass spectrometry system. The UPLC conditions and materials were as follows: column type: Agilent SB-C18 (1.8 µm, 2.1 × 100 mm); solvent system: pure water (0.1% formic acid) and acetonitrile (0.1% formic acid); gradient program: starting at a ratio of 95:5 (v/v) at 0 minutes, transitioning linearly to 5:95 (v/v) over 9 minutes and maintaining this ratio for 1 minute, followed by a return to 95:5 (v/v) over 1.1 minutes and holding for 2.9 minutes; flow rate: 0.35 mL/min; column temperature: 40°C; injection volume: 2 μL. The effluent was connected alternately to an ESI-triple quadrupole-linear ion trap (QTRAP)-MS. The methanol, acetonitrile and formic acid used in this experiment are all chromatographic pure. The ESI source operation parameters were set as follows: source temperature at 500°C; ion spray voltage (IS) at 5500 V (positive ion mode) and −4500 V (negative ion mode); ion source gas I (GSI), gas II(GSII), and curtain gas (CUR) were set at 50, 60, and 25 psi, respectively. Triple quadrupole (QQQ) scans were performed as via multiple reaction monitoring (MRM) experiments with the collision gas (nitrogen) set to medium. For single MRM transitions, declustering potential (DP) and collision energy (CE) were optimized further. A specific set of MRM transitions was monitored for each period according to the metabolites eluted during that time. The qualitative analysis of metabolites was conducted using the proprietary Metware Database (MWDB), while the quantitative analysis was performed using Multiple Reactoin Monitoring (MRM). The specific procedures are as follows: First, the precursor ions of the target substances are screened, followed by the generation of fragment ions through induced ionization. Subsequently, the required characteristic fragment ions are selected to obtain mass spectrometry analysis data for various metabolites. Finally, the peak area of the chromatographic peaks are integrated, and integration corrections are applied.
RNA extraction, transcriptomic sequencing and library construction
The RNA of P. polyphylla was extracted using ethanol precipitation and CTAB-PBIOZOL, and subsequently dissolved in 50 µL of DEPC-treated water. Following extraction, the total RNA was identified and quantified using a Qubit fluorescence quantifier and a Qsep400 high-throughput biofragment analyzer. Oligo (dT) magnetic beads were employed to enrich mRNAs with polyA tails, as most eukaryotic mRNAs possess a polyA tail. Subsequently, a SMARTer PCR cDNA Synthesis Kit was utilized to convert mRNA into cDNA through reverse transcription. Sequencing adapter ligation was performed, followed by DNA magnetic bead purification, and fragment selection was completed to yield a library with insert fragments ranging from 250~350 bp after ligation. The ligated products were then amplified by PCR and purified again using DNA magnetic beads, with the products being solubilized in nuclease-free water. After the initial library construction, concentration detection was performed using a Qubit fluorescence quantifier, followed by fragment size detection using a Qsep400 high-throughput biofragment analyzer. Finally, the effective concentration of the library was accurately quantified using qRT-PCR. Upon passing the library inspection, the full-length transcriptome was sequenced using the PacBio SM-RT Iso-seq platform. The fragments per kilobase of transcript per million fragments mapped (FPKM) value were calculated to determine the gene expression level. To ensure the integrity of the data for subsequent analyses, we utilized fastp to filter out low-quality sequences and adapter sequences. This step was crucial for obtaining clean reads, which serve as the foundation for all further analyses. We then employed SMRTlink software to process the sequence data, resulting in high-quality, polished full-length consensus sequences.
Data analysis
One-way analysis of variance (ANOVA) was conducted using IBM SPSS Statistics version 25.0. Hierarchical cluster analysis (HCA) of samples and metabolites was performed using the R package ComplexHeatmap (version 2.9.4). Unsupervised principal component analysis (PCA) was executed using the statistical function prcomp within the R base package (www.r-project.org, version 4.1.2). Mass spectrometry data are processed by software Analyst 1.6.3.
Data Records
The complete raw metabolomics data set for rhizome tissues of Paris polyphylla var. yunnanensis with different growth years can be accessed on the MetaboLights database under the accession number MTBLS1068326. While the raw transcriptome data information has been submitted to the Genome Sequence Archive (GSA) of National Genome Data Center (NGDC) under the accession number CRA02232627.
Technical Validation
To ensure the reliability of the technical quality of the datasets, each data type adheres to an established methodology for its technical validation. In broadly targeted metabolomics, metabolite quantification is performed using triple quadrupole mass spectrometry in multiple reaction monitoring (MRM) mode (Fig. 2a). For enhanced accuracy and reproducibility in metabolite quantification, the parent ion of the target substance is initially screened using a quadrupole, followed by induced ionization in a collision chamber to form fragment ions. These fragment ions are then filtered through a triple quadrupole to isolate the desired ions corresponding to the target feature. Quality control (QC) samples are utilized to assess the reproducibility of samples processed by the same method, prepared from a mixture of sample extracts. During instrumental analysis, one QC sample is inserted for every ten samples analyzed to monitor the reproducibility of the analytical process and evaluate the consistency of metabolite extraction and detection. The principal component 1 (PC1) scores of the QC sample solutions collected at different times fall within the range of positive and negative standard deviations (Fig. 2b). By overlaying and displaying the analysis of the total ionic current (TIC) plots of the different QC samples (Fig. 3), these results demonstrate high overlap in the total ion current curves detected by metabolites, indicating consistent retention times and peak intensities. This suggests that the signal stability of the mass spectrometer is robust when the same sample is detected at different times, confirming that the instrument’s status was stable and reliable throughout the experiment and that the analytical method was reproducible. Hierarchical clustering analysis of the metabolites from P. polyphylla revealed significant differences in metabolic profiles among samples of varying ages, while samples within the same group exhibited similar metabolic profiles (Fig. 4). It further shows that P. polyphylla has significant differences in different growth years.

(a) Schematic diagram of mass spectrometry multi-reaction monitoring mode. (b) PC1 control chart for all samples.

Detection of TIC overlay by QC sample mass spectrometry. (a) Negative ion mode. (b) Positive ion mode.

Accumulation patterns of metabolite detected from all samples.
During the transcriptome sequencing process, the original third-generation data were processed using the official PacBio software package SMRTlink to obtain consistent transcript sequences. The second-generation data were employed for quality control to eliminate junctions and low-quality sequences, while the third-generation transcripts underwent error correction using the second-generation data and de-redundancy via CD-HIT after obtaining clean data. Subsequent analyses were based on these transcripts. Principal component analysis (PCA), an unsupervised pattern analysis method, elucidates the most prominent features within a multidimensional data matrix. By examining the differences in gene expression across different years of P. polyphylla through PCA, PC1 (20.80%) and PC2 (14.52%) effectively distinguished samples from different years, while samples from the same year clustered together. This indicates a higher reproducibility of the samples and significant differences in gene expression of P. polyphylla across years, consistent with the results from hierarchical clustering analysis (HCA) (Fig. 5).

PCA scores of Unigene detected from all samples.
Code availability
The software used in this study is implemented according to its manual or protocol, and the detailed information of software version and parameters is described in the methods part. This study does not use any custom code for data management or verification.
References
Li, Z. H. & Du, X. W. Research progress and quality marker prediction analysis of Paris polyphylla var. yunnanensis. Chinese Traditional and Herbal Drugs 54, 3032–3048 (2023).
Zhou, D., Pan, Q. D., Yan, X. X., Gao, L. & Yang, L. X. Research progress of steroidal saponins in Paris polyphylla var. yunnanensis and their microbial transformation. China Journal of Chinese Materia Medica 47(18), 4863–4876 (2022).
Jiang, Y. et al. Research progress on chemical constituents,pharmacological actities, and clinical applications of Paris polyphylla var. yunnanensis. Chinese Traditional and Herbal Drugs 53(23), 7633–7648 (2022).
Li, M. et al. Applications of HPLC/MS in the analysis of traditional Chinese medicines. Journal of Pharmaceutical Analysis 1(2), 81–91 (2011).
Peng, S. J. et al. Screening and validation of reference genes for real-time fluorescence quantitative PCR in Paris polyphylla var. yunnanensis and P. forrestii. Plant Physiology Journal 59(05), 997–1007 (2023).
Xu, P. et al. Metabolomics and transcriptomics profiling of three Paris species with varied polyphyllin compositions. Scientific Data 11(1), 1293 (2024).
Zhou, N. et al. Genetic diversity, chemical components, and property of biomass Paris polyphylla var. yunnanensis. Frontiers in bioengineering and biotechnology 9, 713860 (2021).
Chen, S. L. et al. Conservation and sustainable use of medicinal plants: problems, progress, and prospects. Chinese medicine 11, 37 (2016).
Zhang, H., L, Z. Medicinal value and cultivation techniques of Paris polyphylla var. yunnanensis[J]. World Tropical Agriculture Information, (01), 8-9 (2020).
Yang, Z. L., Yang, X. & Wang, L. N. Effects of harvest rotations and growth time on growth and medicinal active ingredient content of Magnolia officinalis. Scientia Silvae Sinicae 53(1), 47–53 (2017).
Gao, H. Y., Shi, M., Zhang, H. J., Shang, H. L., Yang, Q. Integrated metabolomic and transcriptomic analyses revealed metabolite variations and regulatory networks in Cinnamomum cassia Presl from four growth years. Frontiers in Plant Science 14 (2024).
Wang, Z. et al. Evaluation of the quality of codonopsis radix in different growth years by the AHP-CRITIC method. Chemistry and Biodiversity 20(6), e202201108 (2023).
Wang, Y. Z. & Shen, T. ATR-FTIR combined with chemometrics for origin identification of cultivated Paris polyphylla var. yunnanensis. Chinese Traditional and Herbal Drugs 54(05), 1574–1581 (2023).
Wang, Q. et al. Correlation analysis of quality,origin and phenotypic characters of Paris polyphylla var. yunnanensis. China Journal of Chinese Materia Medica 44(15), 3203–3212 (2019).
Wu, Y. Y., Jin, L., Ma, Y. J., Wang, K. L. & Li, H. F. A comparative study on the fingerprint of Paris polyphylla var. Polyphylla and Paris polyphylla var. yunnanensis from different origins in Dali and seven main steroidal saponins by HPLC. Chinese Pharmaceutical Journal 55(11), 875–882 (2020).
Yang, Y. G., Zhang, J., Zhao, Y. L., Zhang, J. Y. & Wang, Y. Z. Quantitative determination and evaluation of Paris polyphylla var. yunnanensis with different harvesting times using UPLC-UV-MS and FT-IR spectroscopy in combination with partial least squares discriminant analysis. Biomedical Chromatography 31(7), e3913 (2017).
Wu, Z., Zhang, J., Xu, F., Wang, Y. & Zhang, J. Rapid and simple determination of polyphyllin I, II, VI, and VII in different harvest times of cultivated Paris polyphylla Smith var. yunnanensis (Franch.) Hand.-Mazz by UPLC-MS/MS and FT-IR. Journal of natural medicines 71(1), 139–147 (2017).
Qubie, J. Z. et al. Identification of genes involved in biosynthesis of spirostane-type polyphyllin in Paris fargesii based on transcriptome analysis. Chinese Traditional and Herbal Drugs 54(20), 6798–6812 (2023).
Gao, X. et al. Transcriptome analysis of Paris polyphylla var. yunnanensis illuminates the biosynthesis and accumulation of steroidal saponins in rhizomes and leaves. Phytochemistry 178, 112460 (2020).
Li, C. W. C. B., Yang, M. & Li, H. F. Research of steroidal saponins active components accumulation in different parts of multiple stems Paris polyphylla Smith var. yunnanensis. Liaoning Journal of Traditional Chinese Medicine 28(10), 2517–2518 (2017).
Wang, L. N. et al. Dynamic accumulation comparison of nine saponins in the rhizomes of six Paris plants with different growth period[J]. Frontiers in Pharmaceutical Sciences 21(12), 2123–2127 (2018).
Kim, S. J., Kim, S. H., Kim, J. H., Hwang, S. & Yoo, H. J. Understanding metabolomics in biomedical research. Endocrinology and Metabolism 31(1), 7 (2016).
Shen, S. Q., Zhan, C. S., Yang, C. K., Fernie, A. R. & Luo, J. Metabolomics-centered mining of plant metabolic diversity and function: Past decade and future perspectives. Molecular Plant 16(1), 43–63 (2023).
Klassen, A. et al. Metabolomics: Definitions and significance in systems biology. Advances in Experimental Medicine and Biology 965, 3–17 (2017).
Liu, C. X. et al. Research progress on the integration of metabolomics and transcriptomics in the metabolism of medicinal plants. Northern Horticulture, 1-7 (2025).
Xiaoye, Z. et al. MTBLS10683: Transcriptomics and metabolomics datasets of Paris polyphylla var. yunnanensis with different growth years. MetaboLights http://www.ebi.ac.uk/metabolights/MTBLS10683. (2025).
NGDC histology raw data archives (CRA022326). https://ngdc.cncb.ac.cn/gsa/browse/CRA022326 (2025).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82160718), the Yunnan Applied Basic Research Project (202101AZ070001-163), High-Level Talents Project of Yunnan Province (YNWR-QNBJ-2020-257, XDYC-QNRC-2022-0277), Innovative Scientific Research Project of “Key support and characteristic of Yunnan first-class discipline-traditional Chines Medicine”, and the open subject of Yunnan Dai and Yi medicines key laboratory (2024SS24060). We thank Wuhan Metware Biotechnology Co., Ltd., for its technical support in the metabolome and transcriptome analysis.
Author information
Authors and Affiliations
Contributions
Xiaoye Zhang: Methodology, Investigation, Data curation, Writing - original draft. Jia Chen: Methodology, Data curation, Investigation. Ping Xu: Methodology, Data curation. Xuan Zhang: Data analysis. Shuiyi Yang: Investigation. Jie Zhang: Data analysis Xiheng Wan: material acquisition. Yingsi Gao: material acquisition. Guowei Zheng: Supervision, Resources, Writing-review and editing, Project administration.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Chen, J., Xu, P. et al. Transcriptomics and metabolomics profiling of Paris polyphylla var. yunnanensis with different growth years. Sci Data 12, 866 (2025). https://doi.org/10.1038/s41597-025-05242-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05242-y
