
Abstract
Rhizoctonia cerealis, the causal agent of sharp eyespot, is a highly destructive pathogen of wheat. Despite its global importance, the genetic and molecular mechanisms underlying virulence of R. cerealis remain poorly understood. R. cerealis is a dikaryotic organism and the haplotype phase has been isolated. Based on the PacBio HiFi, Oxford Nanopore, and Hi-C platforms, we assembled the first high quality telomere-to-telomere (T2T) haplotype-resolved genome of R. cerealis, with sizes of 41.50 and 41.05 Mb, and N50 sizes of 2.67 and 2.42 Mb, respectively. High consensus quality values of 57.75 and 57.09 for the two haplotypes validated the accuracy of the assembly. The assembly achieved R-AQI and S-AQI scores of 92.5 and 100, respectively, both indicating reference-level quality. A total 25,353 protein coding genes were predicted for the two haplotypes with a BUSCO score of 96.7%. The genome assembly will serve as the foundation for further research on allele-specific expression, genetic variation and evolution of R. cerealis.
Similar content being viewed by others


Background & Summary
Rhizoctonia cerealis Van der Hoeven (current teleomorph name: Ceratobasidium cereale Murray & Burpee, within the Basidiomycota phylum), a member of the Rhizoctonia anastomosis group AG-D, subgroup I (AG-DI), is a soilborne basidiomycete that causes sharp eyespot in wheat. This pathogen is widespread in temperate wheat growing regions of Europe, North America, Africa, Oceania, and Asia1. Sharp eyespot has been reported every year in China since 2016, with the affected area approaching 8 million hectares (https://www.natesc.org.cn/). Severe infection leads to death of young tillers, lodging, and appearance of white heads1. The number of resistant varieties among those currently grown is limited, and control strategies primarily depend on the use of fungicides. The identification of pathogenicity genes could provide crucial insights for disease control strategies. However, previous studies were hindered by lack of a high-quality reference genome sequence, which would be beneficial for comprehending the genetic basis of pathogenicity, biochemical mechanisms, and infection processes.
R. cerealis is a dikaryotic fungus; its mycelium is white in color on a PDA agar plate (Fig. 1a), and each hyphal cell contains two nuclei (Fig. 1b). Over time, the inoculation, or infection, site on the wheat seedling leaf sheath and underlying stem changes from white to brown or black (Fig. 1c). Symptoms on wheat leaf sheaths and stems in the field appear as well-defined semicircular or oval lesions with gray-brown centers and a brown periphery (Fig. 1d), hence the name, eyespot. With prolonged infection spikes of infected tillers undergo premature ripening expressed as white heads (Fig. 1e). Constructing a haplotype-resolved genome facilitates identification of genomic variations between homologous chromosomes, such as single nucleotide polymorphisms (SNP) and structureal variations (SV). This enables exploration allele-specific gene expression at different stages of disease development, and elucidation of the impact of dikaryotism on the genome biology.

Morphology of R. cerealis and signs and symptoms of sharp eyespot in wheat. (a) The front and back sides of R. cerealis hyphae on a PDA agar plate at five days post inoculation. (b) All hyphal cells contain two nuclei as shown by DAPI staining. (c) Symptoms on wheat seedling leaf sheaths and stems at 5 dpi. (d) Field Symptoms on wheat leaf sheaths and stems in the field. (e) Symptoms on wheat spikes in the field.
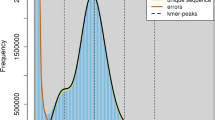
Until now, a chromosome-level genome assembly for R. cerealis has not been reported. Multiple sequencing platforms were used to obtain the present haplotype-resolved, chromosome-level genome assembly of strain R0301. Illumina sequencing yielded 10.65 Gp of clean data (Table 1), which was used for k-mer analysis. The haploid genome of R0301 was estimated to be 40.66 Mb, with a heterozygosity of 1.31% based on a k-mer value of 21 (Fig. 2). The PacBio sequel II platform generated 6.53 Gp of HiFi reads, with 99.9% accuracy and an N50 length of 19,881 bp (Table 1). The Nanopore ONT platform applying ultra-long sequencing technology, yielded an N50 length of 54,315 bp and 16.86 Gp of reads (Table 1), which were highly beneficial for assembling highly repetitive regions, such as centromeres and telomeres, and for generating a gap-free genome. High-throughput chromosome conformation capture (Hi-C) technology provided 17.83 Gb of reads for long-range scaffolding (Table 1). To acquire the telomere-to-telomere (T2T) and haplotype-resolved genome, HiFiasm2 (v.0.19.9) was employed along with the HiFi, ONT, and Hi-C data. Finally, the two haplotypes were separated, and the mitochondrion assembled. Based on Hi-C links, 41.50 Mb of contigs from the A haplotype genome and 41.05 Mb from the B haplotype genome were anchored onto chromosomes (Table 2). The final assembly consisted of 16 chromosome pairs without gaps, achieving a scaffold N50 of 2.67 Mb for haplotype A and 2.42 Mb for haplotype B (Fig. 3, Table 2). Gene annotation on the two haploid genomes identified 12,351 and 12,309 genes for the A and B genomes, respectively (Fig. 3, Table 3). Multiple assessment methods confirmed high continuity, base accuracy, and completeness of the haplotype-resolved assemblies (Table 2). The results of this study will be a useful resource for community research on pathogenicity, genetic variation, and evolution of R. cerealis.

Genome size estimation based on 21-mer distribution. The size of the R0301 genome was estimated to be 40.66 Mb using the frequency distribution of 21-mer and the 1:2 depth ratio between peaks supports a diploid genome with 1.31% heterozygosity.

Circos plot illustrating the genomic features of isolate R0301 in 10 kb windows. Gene density, transposable element (TE) density, GC content, and collinear blocks between the two haplotype genomes (from the outermost to the innermost circles).
Methods
DNA extraction, library construction and sequencing
The CTAB3 method was used for extracting DNA used in Illumina sequencing. Sequencing was performed with an Illumina Novaseq instrument to obtain a PE 150-bp PCR-free library with a 350 bp insert size. High molecular weight (HMW) DNA was extracted following a protocol for BAC library construction4 and then sheared using a Covaris g-TUBE device with 20 kb settings. The sheared DNA was purified and concentrated with AmpureXP beads and subsequently used for single-molecule real-time (SMRT) bell preparation according to the manufacturer’s protocol. HMW DNA, also used for the 1D library, was prepared and sequenced by a Nanopore PromethION sequencer. The same nuclear DNA for Hi-C library construction was cross-linked and then digested with restriction enzyme DpnII, resulting in pairs of distally located but physically interacting DNA molecules. The sticky ends of these digested fragments were biotinylated and ligated to form chimeric circles. Biotinylated circles, chimeras of the physically associated DNA molecules from the cross-linking, were enriched, sheared, and sequenced with the Illumina Novaseq platform.
RNA extraction, library construction and sequencing
RNA was extracted separately from R. cerealis cultured on potato dextrose agar (PDA) medium and from fungal-infected wheat sheaths at 7, 14, and 21 days post-infection (dpi). The extracted RNA was subsequently sequenced using the Illumina Novaseq platform.
Genome size and estimation of heterozygosity
Based on the Illumina reads, Jellyfish (v2.2.10, -m 21)5 was used to count k-mers, and Genomescope (v1.0.0)6 was employed to determine genome size and the heterozygosity level.
Haplotype-resolved genome assembly
HiFi reads, ultra-long reads, and Hi-C reads were assembled into contigs using hifiasm2 (v0.19.9) with parameters -h1 -h2 -ul for haplotype phasing. Clean Hi-C paired-end reads were aligned with the assembly using Juicer (v1.6)7 with the BWA8 algorithm to obtain the interaction matrix. The 3d-DNA (v201008) pipeline9 was applied to reorder the contigs into scaffolds. The position of the contigs was manually adjusted based on the Hi-C heatmaps visualized using JuicerBox10 (v1.11.08). The haplotype-resolved genome was polished using short reads and HiFi reads by NextPolish211 with one iterative round.
Gene prediction and functional annotation of the R. cerealis genome assembly
The Funannotate pipeline (v1.7.4, https://github.com/nextgenusfs/funannotate) was used for genome annotation. Seven steps were executed: (1) Renaming the fasta headers, (2) Sorting scaffolds by length to avoid downstream issues, (3) Soft-masking simple repeats in the genome with tantan, (4) Training gene models with the novaseq and Pacbio RNA-seq data trinity de novo assembly sequences, (5) Performing funannotate prediction with corresponding options after training, (6) Updating the annotation with UTR data and correcting gene models inconsistent with RNA-seq data, and (7) Annotating, which automatically incorporating the interproscan, antismash, phobius results.
Effectors play a crucial role in virulence of plant pathogenic fungi12. A comprehensive pipeline was employed to identify candidate effectors. Firstly, SignalP (v5.0)13 and Phobius (v1.01)14 were used to predict proteins with signal peptides. The proteins predicted by both sofrwares were combined and input into the next step. Then, TMHMM (v2.0c, to remove proteins with ExpAA larger than 18)15, ApoplastP (v1.0.1)16, LOCALIZER (v1.0.4)17, and EffectorP (v2.0)18 were used to predict candidate effectors.
AlleleFinder (https://github.com/sc-zhang/AlleleFinder) was used to identify alleles. Functional gene enrichment analysis was performed using the clusterProfiler package19. GO terms and KEGG pathways with p-adjust < 0.05 and q-value < 0.05 were regarded as significantly enriched. Proteins from the two haplotype genomes of R0301 were compared using BLASTP (-evalue 1e-6 -outfmt 6). Collinear blocks were identified using MCScanX (v0.8)20.
The Funannotate pipeline predicted 24,660 genes (25,353 isoforms). The properties of these gene, such as average CDS length and number of CDS in haplotypes A and B are presented in Table 3. Chromosome 02 (Chr02) exhibited the highest gene density (with an average 0.37 genes per kb), and Chr05 had the lowest gene density (0.20 genes per kb) (Table 4). Among tranlated proteins, 1,781 (7.02%) were annotated as secreted proteins (SPs), and 485 (1.91%) were annotated as cytoplasmic/apoplastic effectors (Supplementary Table S1). A majority of genes (81.3%, 10,582 of 13,130) were bi-allelic, and 18.7% (2,548 of 13,132) were mono-allelic, with 1,732 specifically in haplotype A and 816 in haplotype B (Table 4). Subsequent analysis revealed a significant influence on glucan metabolism and cell surface receptor signaling pathways (Fig. 4, Supplementary Table S2).

Gene ontology (GO) enrichment analysis of isolate R0301 genome specific genes. Significantly enriched terms (p.adjust < 0.05, q-value < 0.05) are categorized into three ontologies: biological processes (BP), cellular components (CC), and molecular functions (MF).
Transposable elements (TEs) were detected and annotated by HiTE21; 17.78% of the genome consisted of TEs. Long terminal repeats (LTRs) and non-LTR retrotransposons were the most abundant TEs, accounting for 14.34% of the two haplotype genomes (Table 5).
Synteny analysis
MUMmer22 was used for whole-genome alignments with the default parameters to investigate the differences between the two haplotypes. The alignment results were filtered using delta-filter with the parameters ‘-m -i 80 -l 100’. After format conversion with the ‘show-coords’ command, SyRI (v1.6.3)23 was employed to detect syntenic regions and structural variations using the default parameters. Plotsr (v1.1.1)24 was used to visualize these variations. A total of 1,443 syntenic regions with a cumulative size of 58.89 Mb (71.34%) was detected (Fig. 5), indicating moderate similarity between the two haplotypes. A total of 427,567 SNPs and 22,409 indels (11,686 insertions and 11,073 deletions) were identified between HapA and HapB. Additionally, 1,240 translocations with a cumulative size of 6.77 Mb (~8.20%), 31 inversions with a cumulative size of 1.36 Mb (~1.64%), and 3,640 duplications with a cumulative size of 8.94 Mb (~10.82%) were detected (Fig. 5).

Comparative genomic landscape of synteny, structural variation, and telomere distribution in the haplotype-resolved genome. The 16 chromosomes belonging to HapA and HapB are shown in dark blue and orange, respectively. Synteny blocks between homoeologous regions are shown in grey. The black triangles indicate the presence of telomere sequence repeats.
Data Records
The PacBio HiFi and assemblies of R. cerealis isolate R0301 generated in this study are available in NCBI under BioProject PRJNA1187694 and PRJNA118790. The Oxford Nanopore, Hi-C, Illumina sequencing reads of R0301 were obtained from the sequencing data deposited in the BioProject PRJNA717151. The PacBio HiFi sequencing reads of R0301 have been deposited in the NCBI Sequence Read Archive database with accession group numbers SRP54689625. The Oxford Nanopore, Hi-C, Illumina sequencing reads of R0301 have been submitted to the NCBI Sequence Read Archive database (SRP31225826). Genome assembly is available from GenBank in the NCBI with accession numbers GCA_049000075.127 and GCA_048999985.128. The genome assembly and gene annotation results were also deposited in the Figshare database29.
Technical Validation
Evaluation of the assembled genome
The quality of the genome assembly was evaluated using multiple methods. (1) Accuracy of the Hi-C based chromosome construction was evaluated by visualizing the chromatin contact matrix and contact maps using JuicerBox (v1.11.08)10. The Hi-C heatmap showed no errors in the two haplotype genomes (Fig. 6). (2) The completeness and duplication score of the haplotype A and B genomes were evaluated by BUSCO (v5.7.1)30 with the “basidiomycota odb9” database. The complete BUSCO scores (including single-copy and duplicated) of the two haplotypes as well as their combined assembly, accounted for 98.9%, 98.7% and 99.1% in genome mode (Table 2), respectively, suggesting good completeness of the genome assembly. (3) The accuracy of the genome was evaluated by mapping the WGS sequencing reads against the genome and calculating the mapping rate and coverage by Qualimap231 and Pandepth32. The mapping rate was over 93.3%, and the coverage rate (>1X) was 100% for all three data types in both the HapA and HapB genomes (Supplementary Table S3). (4) CRAQ33 was used to evaluate the assembly errors of the R0301 genome. It used clipping information by mapping the original sequencing reads againest the de novo genome assembly, defining a reference level Assembly Quality Indicator (AQI over 92.50%) (Table 2). (5) Merqury (v1.1)34 was used to assess k-mer completeness and the consensus quality value of the R0301 genome assemblies based on the 21-mer hybrid Merqury kmer database of Illumina PCR-free reads. The HapA and HapB genomes, as well as their combined assembly, had a quality values (QV) of 57.75, 57.09 and 57.41, respectively (Table 2). (6) The LTR Assembly Index (LAI)35 value method was used to evaluate the assembly level of the genome based on repeat sequences. The LAI values for the genoems were 33.60, 28.75 and 28.69 (Table 2). (7) A total of 50 telomeres (Fig. 5; Table 2; Table 4) were identified across 32 chromosomes in the haplotype-resolved, gap-free genome of R0301 according to the characteristic repetitive base sequence of telomeric regions (CCCTAAA/TTTAGGG). Complete T2T assemblies were achieved for 19 chromosomes, with 7 T2T assemblies for haplotype A and 12 for haplotype B (Fig. 5; Table 2). These results indicate that the R0301 assembly constitutes the first haplotype-resolved, chromosome-scale genome resource for the R. cerealis species, reaching a near-complete standard with high contiguity and base accuracy.

Inter-chromosomal Hi-C contact map of the full dikaryotic genome assembly for isolate R0301 (32 chromosomes). The nuclear HapA and HapB display a clear signal of spatial separation, the blue boxes represent chromosomal regions.
Evaluation of the gene annotation
The quality of the predicted proteins was assessed using BUSCO (v5.7.1)30 with the “basidiomycota odb9” database in protein mode. The complete BUSCO scores for the two haplotypes, as well as their combined assembly, were 94.0%, 94.3% and 96.7%, respectively (Table 2). These results indicated a high level of quality in the gene annotation process.
Code availability
All software and pipelines employed in this study were executed with the parameters detailed in the Methods section. In cases where specific parameters for the software were not provided, default parameters recommended by the developer were utilized. Circos visualizations were generated using ShinyCircos-V2.036.
References
Hamada, M. S., Yin, Y. N., Chen, H. G. & Ma, Z. H. The escalating threat of Rhizoctonia cerealis, the causal agent of sharp eyespot in wheat. Pest Manag. Sci. 67, 1411–1419 (2011).
Cheng, H. Y. et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. 40, 1332–1335 (2022).
Rogstad, S. H. Saturated nacl-ctab solution as a means of field preservation of leaves for DNA analyses. Taxon 41, 701–708 (1992).
Zeng, Q. D. et al. Construction and characterization of a bacterial artificial chromosome library for the hexaploid wheat line 92R137. Biomed Res. Int. 2014, 9 (2014).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Robinson, J. T. et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst. 6, 256–258 (2018).
Hu, J. et al. NextPolish2: a repeat-aware polishing tool for genomes assembled using HiFi long reads. Genom. Proteomics Bioinformatics 22, 6 (2024).
Bai, X. et al. A candidate effector protein PstCFEM1 contributes to virulence of stripe rust fungus and impairs wheat immunity. Stress Biology 2, 21 (2022).
Almagro Armenteros, J. J. et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423 (2019).
Kall, L., Krogh, A. & Sonnhammer, E. L. Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 35, W429–432 (2007).
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Sperschneider, J., Dodds, P. N., Singh, K. B. & Taylor, J. M. ApoplastP: prediction of effectors and plant proteins in the apoplast using machine learning. New Phytol. 217, 1764–1778 (2018).
Sperschneider, J. et al. LOCALIZER: subcellular localization prediction of both plant and effector proteins in the plant cell. Scientific Reports 7, 44598 (2017).
Sperschneider, J., Dodds, P. N., Gardiner, D. M., Singh, K. B. & Taylor, J. M. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol. Plant Pathol. 19, 2094–2110 (2018).
Yu, G. C., Wang, L. G., Han, Y. Y. & He, Q. Y. clusterProfiler: an R Package for comparing biological themes among gene clusters. Omics-a Journal of Integrative Biology 16, 284–287 (2012).
Wang, Y. P. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Hu, K. et al. HiTE: a fast and accurate dynamic boundary adjustment approach for full-length transposable element detection and annotation. Nat. Commun. 15, 5573 (2024).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, 14 (2018).
Goel, M., Sun, H. Q., Jiao, W. B. & Schneeberger, K. SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 20, 13 (2019).
Goel, M. & Schneeberger, K. plotsr: visualizing structural similarities and rearrangements between multiple genomes. Bioinformatics 38, 5328–5328 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP546896 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP312258 (2021).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_049000075.1 (2025).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_048999985.1 (2025).
Zeng, Q. D. Haplotype-resolved T2T genome assembly of the dikaryotic wheat fungus Rhizoctonia cerealis, Figshare https://doi.org/10.6084/m9.figshare.28112558 (2024).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016).
Yu, H. Y., Shi, C. M., He, W. M., Li, F. & Ouyang, B. PanDepth, an ultrafast and efficient genomic tool for coverage calculation. Briefings in Bioinformatics 25, 256–258 (2024).
Li, K. P., Xu, P., Wang, J. P., Yi, X. & Jiao, Y. N. Identification of errors in draft genome assemblies at single-nucleotide resolution for quality assessment and improvement. Nat. Commun. 14, 12 (2023).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 27 (2020).
Ou, S. J., Chen, J. F. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, 11 (2018).
Wang, Y. et al. shinyCircos-V2.0: Leveraging the creation of Circos plot with enhanced usability and advanced features. Imeta 2, e109 (2023).
Acknowledgements
The authors express their gratitude to Prof. R. A. McIntosh, Plant Breeding Institute, University of Sydney for language editing of the draft manuscript. They also extend their thanks to Dr Hua Zhao and Guoliang Pei (State Key Laboratory for Crop Stress Resistance and High-Efficiency Production, Northwest A&F University) for assistance with confocal microscopy and access to the high-performance server. This work was supported by the Natural Science Basic Research Plan in Shaanxi Province of China (2024NC2-GJHX-36, 2020JCW-16), China Agriculture Research System of Ministry of Finance and Ministry of Agriculture and Rural Affairs (CARS-3), grants from the Opening Foundation of the State Key Laboratory for Crop Stress Resistance and High-Efficiency Production (CSBAAKF2018001).
Author information
Authors and Affiliations
Contributions
Qingdong Zeng, Zhensheng Kang and Dejun Han designed and supervised the study. Yu Li, Wei Li, Hao-Hao Yan, Fengping Yuan, Huai-Gu Chen collected and created the experimental materials. Jiangna Han analyzed the data and wrote the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, JN., Li, Y., Li, W. et al. Haplotype-resolved telomere-to-telomere genome assembly of the dikaryotic fungus pathogen Rhizoctonia cerealis. Sci Data 12, 951 (2025). https://doi.org/10.1038/s41597-025-05260-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-025-05260-w
