
Abstract
Based on the dual response of RhB@UiO-67 (1:6) to Cu2+ and Fe3+, a proportional fluorescent probe with (I392/I581) as the output signal was developed to recognize Cu2+ and Fe3+. Developing highly sensitive and selective trace metal ions probes is crucial to human health and ecological sustainability. In this work, a series of ratio fluorescent probes (RhB@UiO-67) were successfully synthesized using a one-pot method to enable fluorescence sensing of Cu2+ and Fe3+ at low concentrations. The proportional fluorescent probe RhB@UiO-67 (1:6) exhibited simultaneous quenching of Cu2+ and Fe3+, which was found to be of interest. Furthermore, the limits of detection (LODs) for Cu2+ and Fe3+ were determined to be 2.76 μM and 0.76 μM, respectively, for RhB@UiO-67 (1:6). These values were significantly superior to those reported for previous sensors, indicating the probe’s effectiveness in detecting Cu2+ and Fe3+ in an ethanol medium. Additionally, RhB@UiO-67 (1:6) demonstrated exceptional immunity and reproducibility towards Cu2+ and Fe3+. The observed fluorescence quenching of Cu2+ and Fe3+ was primarily attributed to the mechanisms of fluorescence resonance energy transfer (FRET), photoinduced electron transfer (PET), and competitive absorption (CA). This work establishes a valuable foundation for the future study and utilization of Cu2+ and Fe3+ sensing technologies.
Similar content being viewed by others
Introduction
Metal elements are essential for biological growth and development, and have significant effects on life systems1. Iron, for example, is extensively involved in oxygen absorption and transportation, hemoglobin formation, gene regulation, and cholesterol-like synthesis. Similarly, copper is crucial for human health and plays a vital role in maintaining biological growth, metabolism, and homeostasis. Nevertheless, an excess or deficiency of Cu2+ or Fe3+ can disrupt the physiological system and result in various diseases, seriously affecting people's lives and health2,3,4. Therefore, it is imperative to develop a technological solution capable of promptly and precisely identifying Cu2+ and Fe3+.
To date, several conventional approaches have been used to analyze trace metal ions, involving inductively coupled plasma mass spectrometry (ICP-MS)5, high-performance liquid chromatography (HPLC)6, atomic absorption spectrometry (ABS)7, and electrochemical analysis8. However, their practical implementation is constrained by their cumbersome operation and expensive equipment. Hence, an imperative requirement arises for a reaction-based technique that is both cost-effective and expeditious in detecting minute quantities of metal ions. Luminescent metal–organic frameworks (LMOFs) have recently garnered considerable interest owing to their numerous advantages, encompassing diverse luminous modes, rapid response, substantial specific surface area, and satisfactory modifiability9. This has resulted in extensive application of LMOFs throughout many fields, including sensing10, gas separation11, catalysis12, and drug transportation13. Particularly noteworthy is the remarkable potential in fluorescence sensing, encompassing the detection of metal ions14, small molecules15, gases16, volatile organic compounds (VOCs)17, and explosives18. For instance, Zhang et al. have effectively prepared a Tb-based MOF (Tb-TATB) to detect Cu2+ and Fe3+ ions, achieving limits of detection (LODs) of 2.92 μM and 4.84 μM, respectively19. In our previous work, an imidazole-based material (Ag/Zn-ZIF-8 (1:1)) was synthesized for the detection of Cu2+ and Fe3+, and the LODs reached 6.7 μM and 3.9 μM, respectively20. However, the efficacy of these fluorescent probes in detecting Cu2+ and Fe3+ was found to be unsatisfactory due to their reliance on the change in single-emission fluorescence intensity for sensing, which was often influenced by uncontrollable variables like photobleaching, light scattering, and concentration inhomogeneity21. Ratiometric fluorescent probes could effectively overcome these drawbacks due to their multi-occurrence self-calibration signals. Considering the excellent resultant stability and unique optical properties of Zr-MOF, combining Zr-MOF with fluorescent dyes is regarded as an intuitive and effective method, which not only restricts the migration and aggregation of fluorescent dye molecules by Zr-MOF but also generates peculiar fluorescent properties through the subject-object interaction22.
Based on the above factors, a series of ratiometric fluorescent probes were successfully prepared in this work by encapsulating rhodamine B (RhB) into UiO-67. In addition, Cu2+ and Fe3+, as hard acids, can easily coordinate with hard bases such as carboxyl groups23,24. The organic ligands biphenyl-4,4′-dicarboxylate (H2BPDC) and RhB have abundant carboxyl groups, which can easily coordinate with each other and Cu2+ and Fe3+, significantly improving the selectivity and detection limits of RhB@UiO-67 for Cu2+ and Fe3+25. The LODs of Cu2+ and Fe3+ for RhB@UiO-67 were 2.76 μM and 0.76 μM, respectively. Additionally, the probe was also shown to have excellent potential for sensing Cu2+ and Fe3+ in terms of its sensitivity, selectivity, immunity to interference as well as reproducible performance. Furthermore, possible sensing mechanisms have been systematically investigated. Consequently, this current study provides a straightforward and effective strategy for exploring the efficient sensing of trace metal ions (Cu2+ and Fe3+).
Experimental
Preparation of LMOFs
UiO-67 was prepared with adjustments based on previous literature26. ZrCl4 (0.5 mmoL, 116.52 mg), and H2BPDC (0.5 mmoL, 121.11 mg) were dissolved in 30 mL of DMF and subjected to sonication for 30 min to dissolve. Then, the resulting solution was transferred to a Teflon jar and heated at 120 ℃ for 24 h. The strip samples were subsequently cooled to room temperature and washed three times each with DMF and methanol. The white powder obtained was dried at 120 °C for 48 h, followed by further characterizations. Within this experimental procedure, a series of RhB@UiO-67 were synthesized with varying RhB/H2BPDC ratios of 1:2, 1:4, 1:6, 1:8, and 1:10, respectively. Additionally, the pore structure characteristics of these materials were assessed to ensure maximum surface area and pore volume.
Fluorescence sensing experiments
The synthesized fluorescent probes were activated under vacuum at 120 °C for 12 h for fluorescence sensing. The fluorescence properties of UiO-67 and RhB@UiO-67 (1:6) in different solvents were investigated using a spectrophotometer (Hitachi F-7100) with excitation/emission slits of 5.0 nm and photomultiplier voltage of 300 V. The concentration of MOFs was 0.33 mg/mL in selectivity, titration, and immunity experiments.
Density functional theory (DFT) calculation
In this experiment, geometry optimization, the energy levels of lowest unoccupied molecular orbitals (LUMOs) and highest occupied molecular orbitals (HOMOs) were evaluated using density functional theory (DFT) and optimized using Dmol3 of the Materials Studio 2019 package. The all-electron interaction theory (AER) potential accounts for the interaction between electrons and ions. All atoms were allowed to spin unrestricted during the structure optimization process. The GGA-PBE functional and DNP4.4 basis sets were used for the calculations. In the current study, the convergence criterion of the electronic self-consistent field (SCF) loop was set to be 10–6 with energy 10–5 Ha, force constant 0.002 Ha/Å, displacement 0.005 Å, and value of smearing 0.05 Ha27,28. Considering that the metal ions might interact with the carboxyl groups in the organic ligands, H2BPDC and RhB, to simplify the computational process, the optimized structure considered only a single carboxyl group and a metal ion on the ligand. All calculations were performed using a solvent model (ethanol) to obtain more accurate and reliable results.
Analysis of real samples
First of all, 100 mg of Alisma plantago-aquatica L. was soaked in 50 mL of ethanol for 24 h and then filtered using a 0.22 μm filter membrane. Eventually, the samples were analyzed with the addition of different concentrations of Cu2+ and Fe3+ solutions (1 μM, 5 μM, and 10 μM) for three times.
Results and discussion
Characterization of RhB@UiO-67
The N2 adsorption–desorption isotherms of obtained RhB@UiO-67 materials at 77 K were presented in Fig. 1a. Obviously, the adsorption capacity of N2 exhibits a rapid increase at lower relative pressures (p/p0), conforming to a typical type I isotherm, indicating the presence of numerous micropores in RhB@UiO-6729. Additionally, the NLDFT pore size distributions of RhB@UiO-67 materials were more uniform compared with those of UiO-67. And, in RhB@UiO-67, the BET surface areas were relatively small, possibly because RhB plugged the pore channels of UiO-67 (Fig. 1b). Meanwhile, the BET surface area increased followed by a decrease with an increase of RhB, reaching a maximum at RhB/H2BPDC of 1:6 (Table 1). This increase may be caused by the presence of new pore structure shapes, and the decrease may be caused by the continuous addition of RhB to disrupt the symmetry of the original UiO-67 leading to partial structural collapse and blockage of the pore channels22,23. RhB@UiO-67 (1:6) was chosen as a potential probe material for metal ion detection due to its significant specific surface area and pore volume.

(a) N2 adsorption–desorption isotherms at 77 K. (b) NLDFT pore size distributions. (c) XPS spectra. (d) FTIR spectra between 4000 and 400 cm−1 of RhB@UiO-67 materials.
Several approaches were used to characterize the RhB@UiO-67. As shown in Fig. S1, SEM–EDS mapping of RhB@UiO-67 revealed that it had a uniform particle size and nanoscale structure, which facilitated a homogeneous dispersion in subsequent sensing experiments. In addition, compared with UiO-67, RhB@UiO-67 (1:6) exhibited a unique element N from RhB (Fig. S1), which indicated that RhB@UiO-67 (1:6) was successfully prepared. Interestingly, the XPS spectrum of RhB@UiO-67 (1:6) also demonstrated a peak at N 1s that was not present in UiO-67, which also proved that RhB@UiO-67 was successfully prepared (Fig. 1c)30.
The PXRD patterns of RhB@UiO-67 materials were very similar to that of pure UiO-67, indicating the well-maintained structure of UiO-67 after the introduction of RhB (Fig. S2). Therefore, it was likely that RhB was encapsulated within the voids rather than physically adsorbed on the surface of UiO-67. In addition, Fig. 1d demonstrated that the FTIR spectra of UiO-67 and RhB@UiO-67 were highly similar, confirming the encapsulation of RhB and the negligible effect of host–guest encapsulation on the structural integrity of UiO-6731. Furthermore, its exceptional thermal stability was demonstrated by its unchanged state at temperatures up to 450 °C (Fig. S3).
Photoluminescence properties
Fluorescence performance of RhB@UiO-67
The solid-state fluorescence properties of the organic ligand (H2BPDC), vacuum-activated UiO-67, and RhB@UiO-67 were examined. The presence of abundant aromatic rings in the π-π conjugated system of the organic ligand resulted in a strong emission at 391 nm upon excitation at 322 nm but the fluorescence intensities of both UiO-67 and RhB@UiO-67 were weaker than that of H2BPDC due to the aggregation-induced quenching (Fig. S4a). A new fluorescence peak appeared at 582 nm when RhB was introduced. In addition, the fluorescence intensity of RhB@UiO-67 ligand out showed an initial enhancement followed by a weakening trend after the addition of RhB (Fig. S4b). The initial enhancement could be caused by the interaction between RhB and UiO-67. In contrast, the subsequent decrease resulted from the fluorescence quenching of RhB due to aggregation induction.
Additionally, the fluorescence behavior of RhB@UiO-67 (1:6) in various solvents (water, MeOH, EtOH, and DMF) was investigated at room temperature. It was evident that RhB@UiO-67 (1:6) demonstrated the best fluorescence behavior in ethanol (Fig S5a). Furthermore, the investigation of the fluorescence stability of RhB@UiO-67 (1:6) involved an examination of its relative fluorescence intensity at different time intervals and pH levels. The results demonstrated that the relative fluorescence intensity (I392/I581) of RhB@UiO-67 (1:6) remained consistent even after being immersed in ethanol for a duration of one week (Fig. S6c). Additionally, the relative fluorescence intensity of RhB@UiO-67 (1:6) showed minimal variation within the pH range of 3 to 10 (Fig. S6b). Afterward, the I392/I581 ratio of RhB@UiO-67 (1:6) was evaluated at different concentrations. Notably, the I392/I581 ratio initially increased with the concentration of RhB@UiO-67 (1:6), reaching its peak at 0.33 mg/mL (Fig. S5), before subsequently declining. Moreover, it was observed that the I392/I581 ratio remained essentially stationary at 60 s (Fig. S6a). Thus, the optimal concentration and incubation time for RhB@UiO-67 (1:6) were 0.33 mg/mL and 60 s, respectively.
Fluorescence detection of Cu2+ and Fe3+
To investigate the impact of RhB doping on the fluorescence properties of UiO-67, photoluminescence sensing experiments were conducted on a range of metal ions. Figure 2a illustrates that RhB@UiO-67 (1:6) presents remarkable selectivity towards Cu2+ and Fe3+ in comparison to other metal ions. The relative fluorescence intensity (Iligand/IRhB) of RhB@UiO-67 (1:6) exhibited a 64.4% increase for Cu2+ and an 84.1% decrease for Fe3+. However, when examining other potential interfering metal ions, the relative fluorescence intensity (Iligand/IRhB) of RhB@UiO-67 (1:6) did not change significantly compared to the blank solution (Fig. 2). Additionally, the fluorescence intensity of RhB@UiO-67 (1:6) experienced a decrease across all ligand wavelengths during the detection of selected metal ions (Cu2+ and Fe3+) in comparison to UiO-67 (Figs. 2a and S7a). Nevertheless, RhB@UiO-67 (1:6) exhibited a noteworthy increase in relative fluorescence intensity (Iligand/IRhB) compared to the blank solution when detecting Cu2+, which may be attributed to the interaction between Cu2+ and H2BPDC/ RhB. In addition, it is evident that the fluorescence intensity of RhB@UiO-67(1:6) for simultaneous Cu2+/Fe3+ sensing is lower than that for Cu2+ sensing, yet higher than that for Fe3+ sensing (Fig. S7a). This discrepancy may be attributed to the competition for energy transfer from RhB@UiO-67(1:6) to Cu2+ or Fe3+ in the context of simultaneous Cu2+/Fe3+ sensing.

(a) Fluorescence responses of RhB@UiO-67 (1:6) (λex = 335 nm) (0.33 mg/mL) in the presence of various metal ions. (b) Relative fluorescence intensity of RhB@UiO-67 (1:6) toward various metal ions in ethanol solution.
The assessment of sensitivity plays a crucial role in the practical application of probes. To evaluate the sensitivity of RhB@UiO-67 (1:6) towards Cu2+ and Fe3+, fluorescence titration experiments were performed. The results indicated that RhB@UiO-67 (1:6) only displayed fluorescence behavior at 392 nm when detecting Cu2+ and Fe3+. Upon the introduction of RhB, RhB@UiO-67 (1:6) exhibited a new peak at 581 nm. Moreover, the fluorescence intensity of UiO-67 and RhB@UiO-67 (1:6) at 392 nm decreased progressively with the increase of Cu2+. Additionally, the fluorescence intensity of RhB@UiO-67 (1:6) at 581 nm also showed a similar trend, indicating an interaction between Cu2+ and the organic ligands H2BPDC and RhB (Figs. 3a and S8a). These observed fluorescence behaviors provided a valuable reference for internal calibration, effectively reducing interference in Cu2+ detection. Therefore, the ratio I392/I581 was deemed a self-calibrated signal ratio for RhB@UiO-67 (1:6). The Cu2+ concentration showed a strong linear correlation with the relative fluorescence value (I392/I581), as represented by the equation I392/I581 = 0.0317X + 5.43917 (X = 0–10 μM), with a high correlation coefficient (R2) of 0.99376. The LOD of Cu2+ by this probe was determined to be 2.76 μM (Fig. 3b), which was significantly lower than that of UiO-67 (17.63 μM) (Fig. S8b). In addition, the chromaticity coordinates (CIE coordinates) of RhB@UiO-67 (1:6) were determined based on the fluorescence spectra, revealing a color transition from pink to light purple with increasing Cu2+ concentration (inset of Fig. 3b).

Fluorescence spectra and linear plot of RhB@UiO-67 (1:6) (λex = 335 nm) in the presence of various concentrations of Cu2+/Fe3+ in ethanol solution (a) and (b), Cu2+; (c) and (d), Fe3+.
The titration experiments conducted on RhB@UiO-67 (1:6) with Fe3+ demonstrated a concentration-dependent relationship in the emission spectra following excitation at 335 nm. The fluorescence intensity of RhB@UiO-67 (1:6) gradually decreased with increasing Fe3+ concentration (Fig. 3c), and the relative fluorescence intensity (I392/I581) of RhB@UiO-67 (1:6) strongly correlated with concentration. Notably, a significant linear correlation was observed when plotting the working curve of relative fluorescence values (I392/I581) against Fe3+ concentration: I392/I581 = − 0.11484X + 5.6225 (X = 0–5 μM), with a correlation coefficient (R2) of 0.99196. The LOD for RhB@UiO-67 toward Fe3+ was estimated to be 0.76 μM (Fig. 3d), which was significantly lower than that of the UiO-67 (15.62 μM) (Fig. S8d). Furthermore, the CIE coordinates of RhB@UiO-67 (1:6) revealed a transition in color from deep red to light red when the concentration of Fe3+ increased (inset of Fig. 3d).
In view of the above, it can be concluded that RhB@UiO-67 (1:6) possesses the potential to effectively monitor the Cu2+ and Fe3+ aspects through the utilization of a multi-response sensing model. Interestingly, RhB@UiO-67 (1:6) exhibited superior sensitivity compared to other probes designed to detect Cu2+ and Fe3+ (Table S1).
The probe must possess the capacity to detect the target analyte when other interfering ions are present. Consequently, anti-interference experiments were performed on RhB@UiO-67 (1:6). As shown in Fig. 4a and b, it is evident that the addition of Cu2+ and Fe3+ resulted in the quenching of RhB@UiO-67 (1:6) when coexisting with other metal ions. This finding suggests that RhB@UiO-67 (1:6) is less affected by other interfering ions when sensing Cu2+ and Fe3+. As a result, RhB@UiO-67 (1:6) shows promise for selectively sensing Cu2+ and Fe3+ in practical applications.

Relative fluorescence intensity of RhB@UiO-67 (1:6) upon the addition of (a) Cu2+ and (b) Fe3+ (10–4 mol/L) when various cations are present (10–4 mol/L) in ethanol; Regeneration properties of RhB@UiO-67 (1:6) after 8 cycles for (c) Cu2+and (d) Fe3+.
The ability to regenerate the probe material is also crucial in evaluating its actual sensing performance. To assess the regeneration capability, the probe material was washed with ethanol, followed by centrifugation and drying before conducting subsequent sensing experiments. The results presented in Fig. 4c and d demonstrate that the relative fluorescence intensity of RhB@UiO-67 (1:6) returned to its initial value after several consecutive sensing experiments. Furthermore, the pore structure of RhB@UiO-67 (1:6) remained essentially unchanged following 5 cycles of regeneration in Cu2+ and Fe3+ ion solutions, as depicted in Fig. S9a,b and Table S2. However, the BET-specific surface area of RhB@UiO-67 (1:6) began to decrease after 8 cycles, potentially due to the degradation of pore channels by Cu2+ or Fe3+ ions after 8 cycles, as shown in Fig. S9c,d and Table S2. These results indicate that the potential reusability of RhB@UiO-67 (1:6) in detecting Cu2+ and Fe3+ ions.
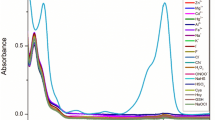
To further understand the fluorescence quenching mechanisms toward Cu2+ and Fe3+, several experiments were conducted. First of all, Fig. 5 depicts the UV absorption profiles of the metal ions under investigation and the fluorescence curves of RhB@UiO-67 (1:6). It was found that among the metal ions under investigation, only the UV absorption profiles of Cu2+ and Fe3+ exhibited overlaps with the fluorescence excitation spectrum of RhB@UiO-67 (1:6), signifying the presence of competitive absorption (CA) between RhB@UiO-67 (1:6) and Cu2+/Fe3+32. Furthermore, the UV absorption profiles of Cu2+ and Fe3+ partially coincided with the fluorescence emission spectrum of RhB@UiO-67 (1:6), suggesting the occurrence of energy transfer between RhB@UiO-67 (1:6) and Cu2+/Fe3+. Moreover, the titration detection of Cu2+/Fe3+ experiments indicated that RhB@UiO-67 (1:6) functioned as a turn-off ratiometric fluorescent probe for both ions. This was evidenced by the observed energy transfer from RhB@UiO-67 (1:6) to Cu2+/Fe3+33. Additionally, the fluorescence lifetimes of RhB@UiO-67 (1:6) at different wavelengths were altered upon the introduction of different concentrations of Cu2+ and Fe3+ ions, demonstrating quenching mechanisms between RhB@UiO-67 (1:6) and Cu2+/Fe3+. The energy transfer efficiencies were determined through calculations based on Fig. S12 (Tables S3, S4)34. Thus, there was fluorescence resonance energy transfer (FRET) in the energy response between RhB@UiO-67 (1:6) and Cu2+/Fe3+.

The UV–vis absorbance spectrum of Cu2+/Fe3+ (10–4 mol/L) and the photoluminescence spectra of RhB@UiO-67 (1:6). (a) Cu2+, (b) Fe3+. (c) UV–vis absorbance spectra of different cations in ethanol solution (10–4 mol/L) and the photoluminescence spectra of RhB@UiO-67 (1:6). (d) Frontal molecular orbital distributions of H2BPDC, Cu2+-H2BPDC, and Fe3+-H2BPDC calculated by DMol3.
Subsequently, FTIR analysis revealed the presence of a peak at 1650 cm–1, which can be attributed to the carboxylate stretching vibration. There was a blue shift of the peak at 1699 cm–1 owing to the interaction between Cu2+/Fe3+ ions and the carboxyl groups present in the organic ligand H2BPDC and RhB (Fig. S10)35,36. Furthermore, the XPS spectra revealed the presence of additional peaks, namely the Cu 2p peak at 935.55 eV, when RhB@UiO-67 (1:6) interacted with Cu2+ ions, and the Fe 2p peak at 722.13 eV, when RhB@UiO-67 (1:6) interacted with Fe3+ ions, which were absent in the RhB@UiO-67 (1:6) material (Fig. S11). Moreover, the high-resolution XPS spectrum of the O 1 s orbital exhibited a minimal change in the binding energy of the -OH bond and a minor variation in the metal-O (metal as Cu/Fe) bond after the interaction with Cu2+ and Fe3+. This observation suggests that Cu2+/Fe3+ ions interact with the carboxyl groups in the organic ligands H2BPDC and RhB37.
To further substantiate this proposed mechanism, the energies of LUMOs and HOMOs were calculated. The LUMOs of both Cu2+-H2BPDC and Fe3+-H2BPDC were found to be lower than that of the ligand, suggesting the occurrence of photoelectron transfer (PET) between RhB@UiO-67 (1:6) and Cu2+/Fe3+ (Fig. 5d)38.
Real samples detection
The reliability and adaptability of the method were further evaluated by the determination of Cu2+ and Fe3+ in Alisma plantago-aquatica L (one Chinese herbal medicine). The standard addition method was employed, and the results, presented in Table 2, exhibited recoveries ranging from 100.8 to 107.8% with the RSD below 2.73% (n = 3). These findings indicate that the method in the study serves as a robust platform for the detection of Cu2+ and Fe3+.
Conclusion
The ratiometric fluorescent probe RhB@UiO-67 prepared by the one-pot method was used for sensing trace amounts of Cu2+ and Fe3+ due to its unique fluorescence properties. Interestingly, RhB@UiO-67 (1:6) exhibited fluorescence behavior with fluorescence quenching for both Cu2+ and Fe3+. Furthermore, the RhB@UiO-67 (1:6) composite demonstrated exceptional immunity, sensitivity, and selectivity towards Cu2+ and Fe3+ ions, with LODs of 2.76 μM and 0.76 μM, respectively. These LODs were significantly lower than those reported in previous studies. Additionally, a comprehensive investigation was conducted to elucidate the potential photoluminescence mechanisms, which revealed that the interaction between the carboxyl group in the framework (acting as Lewis base site) and the metal ions Cu2+/ Fe3+ Lewis acid sites) may be responsible for competitive absorption (CA), resonance energy transfer (FRET), and photon electron transfer (PET) mechanisms. What's more, combining organic dyes with MOFs with stabilized structures is a very promising method for the recognition of Cu2+ and Fe3+, which paves the way for future investigation and practical applications in the detection of multicomponent metal ions.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Xu, L. et al. Nanoenabled intracellular metal ion homeostasis regulation for tumor therapy. Adv. Sci. 11, 2306203–2306224. https://doi.org/10.1002/advs.202306203 (2023).
Geng, R. et al. Bimetallic Cd/Zr-UiO-66 material as a turn-on/off probe for As5+/Fe3+ in organic media. Chemosphere 291, 132827. https://doi.org/10.1016/j.chemosphere.2021.132827 (2022).
Lei, Y. et al. Synthesis of strong magnetic response ZIF-67 for rapid adsorption of Cu2+. Front. Chem. 11, 1135193. https://doi.org/10.3389/fchem.2023.1135193 (2023).
Tian, X. M. et al. Turn-on luminescent sensor toward Fe3+, Cr3+, and Al3+ based on a Co(II) metal-organic framework with open functional sites. Inorg. Chem. 59(5), 2803–2810. https://doi.org/10.1021/acs.inorgchem.9b03152 (2020).
Li, X. M. et al. Discrimination of Pb-Zn deposit types using sphalerite geochemistry: New insights from machine learning algorithm. Geosci. Front. 14, 101580. https://doi.org/10.1016/j.gsf.2023.101580.doi:10.1016/j.gsf.2023.101580 (2023).
Rekhi, H., Rani, S., Sharma, N. & Malik, A. K. A review on recent applications of high-performance liquid chromatography in metal determination and speciation analysis. Crit. Rev. Anal. Chem. 47, 524–537. https://doi.org/10.1080/10408347.2017.1343659 (2017).
Soylak, M., Uzcan, F., Goktas, O. & Gumus, Z. P. Fe3O4-SiO2-MIL-53 (Fe) nanocomposite for magnetic dispersive micro-solid phase extraction of cadmium (II) at trace levels prior to HR-CS-FAAS detection. Food Chem. 429, 136855. https://doi.org/10.1016/j.foodchem.2023.136855 (2023).
Ma, X. et al. Electrochemical sensor based on N-CQDs/AgNPs/β-CD nanomaterials: Application to simultaneous selective determination of Fe(II) and Fe(III) irons released from iron supplement in simulated gastric fluid. Talanta 253, 123959. https://doi.org/10.1016/j.talanta.2022.123959 (2023).
Zhao, Y., Zeng, H., Zhu, X. W., Lu, W. & Li, D. Metal-organic frameworks as photoluminescent biosensing platforms: Mechanisms and applications. Chem. Soc. Rev. 50(7), 4484–4513. https://doi.org/10.1039/d0cs00955e (2021).
Yan, F. et al. Sensing performance and mechanism of carbon dots encapsulated into metal-organic frameworks. Microchim. Acta 189(10), 379. https://doi.org/10.1007/s00604-022-05481-5 (2022).
Fan, W., Zhang, X., Kang, Z., Liu, X. & Sun, D. Isoreticular chemistry within metal-organic frameworks for gas storage and separation. Coord. Chem. Rev. 443, 213968. https://doi.org/10.1016/j.ccr.2021.213968 (2021).
Wang, Q. & Astruc, D. State of the art and prospects in metal-organic framework (MOF)-based and MOF-derived nanocatalysis. Chem. Rev. 120(2), 1438–1511. https://doi.org/10.1021/acs.chemrev.9b00223 (2019).
Zhao, F. et al. Redox homeostasis disruptors based on metal-phenolic network nanoparticles for chemo/chemodynamic synergistic tumor therapy through activating apoptosis and cuproptosis. Adv. Healthc. Mater. 12(29), 2301346. https://doi.org/10.1002/adhm.202301346 (2023).
Wu, L. et al. Highly selective and turn-on fluorescence probe with red shift emission for naked-eye detecting Al3+ and Ga3+ based on metal-organic framework. Chin. Chem. Lett. 33(1), 541–546. https://doi.org/10.1016/j.cclet.2021.06.009 (2022).
Chakraborty, G., Park, I. H., Medishetty, R. & Vittal, J. J. Two-dimensional metal-organic framework materials: Synthesis, structures, properties and applications. Chem. Rev. 121(7), 3751–3891. https://doi.org/10.1021/acs.chemrev.0c01049 (2021).
Li, H. Y., Zhao, S. N., Zang, S. Q. & Li, J. Functional metal-organic frameworks as effective sensors of gases and volatile compounds. Chem. Soc. Rev. 49(17), 6364–6401. https://doi.org/10.1039/c9cs00778d (2020).
Liu, L., Ru, L., Tang, H., Zhang, Z. & Feng, W. A multi-responsive Tb-doped MOF probe for highly specific breath volatile biomarker recognition of lung cancer. J. Mater. Chem. C. 11(8), 3059–3069. https://doi.org/10.1039/d2tc05140k (2023).
Yang, L. et al. A built-in self-calibrating luminescence sensor based on RhB@Zr-MOF for detection of cations, nitro explosives and pesticides. RSC. Adv. 10(33), 19149–19156. https://doi.org/10.1039/d0ra02843f (2020).
Hang, X. et al. A microporous Tb-based MOF for multifunctional detection of the α-CHC, Cu2+, and Fe3+. J. Solid. State Chem. 312, 123232. https://doi.org/10.1016/j.jssc.2022.123232 (2022).
Geng, R. et al. Bimetallic Ag/Zn-ZIF-8: An efficient and sensitive probe for Fe3+ and Cu2+ detection. Colloids Surf. A Physicochem. Eng. Asp. 632, 127755. https://doi.org/10.1016/j.colsurfa.2021.127755 (2022).
Yin, H. Q. & Yin, X. B. Multi-emission from single metal-organic frameworks under single excitation. Small 18(14), 2106587. https://doi.org/10.1002/smll.202106587 (2021).
Wen, X., Xin, C., Zhang, W., Ding, C. & Li, Z. Luminescent dye molecules-embedded zirconium-based metal-organic framework as a bifunctional dual-ratiometric responsive sensor for Cu2+/ Fe3+. J. Lumin. 263, 120092. https://doi.org/10.1016/j.jlumin.2023.120092 (2023).
Zheng, X. et al. Dual-emission Zr-MOF-based composite material as a fluorescence turn-on sensor for the ultrasensitive detection of Al3+. Inorg. Chem. 59(24), 18205–18213. https://doi.org/10.1021/acs.inorgchem.0c02674 (2020).
Cai, D. G. et al. Fabrication and DFT calculation of amine-functionalized metal-organic framework as a turn-on fluorescence sensor for Fe3+ and Al3+ ions. Inorg. Chem. 61(37), 14770–14777. https://doi.org/10.1021/acs.inorgchem.2c02195 (2022).
Li, J. et al. A benzothiadiazole-based Eu3+ metal-organic framework as the turn-on luminescent sensor toward Al3+ and Ga3+ with potential bioimaging application. Inorg. Chem. 61(8), 3607–3615. https://doi.org/10.1021/acs.inorgchem.1c03661 (2022).
Katz, M. J. et al. A facile synthesis of UiO-66, UiO-67 and their derivatives. Chem. Commun. 49(82), 130822. https://doi.org/10.1039/c3cc46105j (2013).
Mei, D. & Yan, B. Flumequine-mediated fluorescent zeolitic imidazolate framework functionalized by Eu3+ for sensitive and selective detection of UO22+, Ni2+, and Cu2+ in nuclear wastewater. J. Hazard. Mater. 447, 130822. https://doi.org/10.1016/j.jhazmat.2023.130822 (2023).
Lei, M., Zhang, Y. C., Wang, M. Y., Yang, W. J. & Gao, Z. Y. Density functional theory investigation of As4, As2, and AsH3 adsorption on Ti-doped graphene. Chem. Eng. J. 421, 129747. https://doi.org/10.1016/j.cej.2021.129747 (2021).
Guo, L. et al. The role of l-histidine as molecular tongs: A strategy of grasping Tb3+ using ZIF-8 to design sensors for monitoring an anthrax biomarker on-the-spot. Chem. Sci. 11(9), 2407–2413. https://doi.org/10.1039/d0sc00030b (2020).
Jin, Y., Lu, H. & Yan, B. A multivariate luminescent MOF based on dye covalent modification serving as a sensitive sensor for Cr2O72−, CrO42− anions and its applications. Dyes Pigm. 194, 109588. https://doi.org/10.1016/j.dyepig.2021.109588 (2021).
Guo, L. et al. A metal-organic framework as selectivity regulator for Fe3+and ascorbic acid detection. Anal. Chem. 91(19), 12453–12460. https://doi.org/10.1021/acs.analchem.9b03143 (2019).
Rath, B. B. & Vittal, J. J. Water stable Zn(II) metal-organic framework as a selective and sensitive luminescent probe for Fe(III) and chromate ions. Inorg. Chem. 59(13), 8818–8826. https://doi.org/10.1021/acs.inorgchem.0c00545 (2020).
Feng, S. et al. A ratiometric fluorescent sensor based on g-CNQDs@Zn-MOF for the sensitive detection of riboflavin via FRET. Spectrochim. Acta A Mol. Biomol. Spectrosc. 246, 119004. https://doi.org/10.1016/j.saa.2020.119004 (2021).
Xiong, J. et al. Aptasensor-based assay for dual-readout determination of aflatoxin B1 in corn and wheat via an electrostatic force–mediated FRET strategy. Microchim. Acta 190(2), 80. https://doi.org/10.1007/s00604-023-05641-1 (2023).
Peng, L. et al. A novel dual emission ratiometric fluorescence sensor Eu3+/CDs@UiO-66 to achieve Cu2+ detection in water environment. Colloids Surf. A Physicochem. Eng. Asp. 664, 131205. https://doi.org/10.1016/j.colsurfa.2023.131205 (2023).
Lv, S. W. et al. A novel and universal metal-organic frameworks sensing platform for selective detection and efficient removal of heavy metal ions. Chem. Eng. J. 375, 122111. https://doi.org/10.1016/j.cej.2019.122111 (2019).
Qiang, Y. et al. UiO-67 decorated on porous carbon derived from Ce-MOF for the enrichment and fluorescence determination of glyphosate. Microchim. Acta 189(3), 130. https://doi.org/10.1007/s00604-022-05236-2 (2022).
Wang, X. et al. Tris(bipyridine)ruthenium(II)-functionalized metal-organic frameworks for the ratiometric fluorescence determination of aluminum ions. Microchim. Acta 189(11), 402. https://doi.org/10.1007/s00604-022-05504-1 (2022).
Funding
This work is sponsored by the Natural Science Foundation of Henan Province (Grant 242300420197), the National Natural Science Foundation of China (Grant 21878122, 82304734), China Postdoctoral Science Foundation (2021M690937), and Zhongjing Core Scholar’s Research Initial Fund of Henan University of Chinese Medicine.
Author information
Authors and Affiliations
Contributions
Teng Zhang: Investigation, Writing-original draft. Rui Cao: Formal analysis and Investigation. Jingying Li: Investigation. Hanxiao Tang: Validation, Funding acquisition. Hang Su: Methodology. Weisheng Feng: Supervision. Zhijuan Zhang: Supervision, Writing-review & editing, Funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, T., Cao, R., Li, J. et al. A dual-responsive RhB-doped MOF probe for simultaneous recognition of Cu2+ and Fe3+. Sci Rep 14, 11740 (2024). https://doi.org/10.1038/s41598-024-62177-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-62177-x
