
Abstract
To explore the hub comorbidity genes and potential pathogenic mechanisms of hypopharyngeal carcinoma with esophageal carcinoma, and evaluate their diagnostic value for hypopharyngeal carcinoma with co-morbid esophageal carcinoma. We performed gene sequencing on tumor tissues from 6 patients with hypopharyngeal squamous cell carcinoma with esophageal squamous cell carcinoma (hereafter referred to as “group A”) and 6 patients with pure hypopharyngeal squamous cell carcinoma (hereafter referred to as “group B”). We analyzed the mechanism of hub genes in the development and progression of hypopharyngeal squamous cell carcinoma with esophageal squamous cell carcinoma through bioinformatics, and constructed an ROC curve and Nomogram prediction model to analyze the value of hub genes in clinical diagnosis and treatment. 44,876 genes were sequenced in 6 patients with group A and 6 patients with group B. Among them, 76 genes showed significant statistical differences between the group A and the group B.47 genes were expressed lower in the group A than in the group B, and 29 genes were expressed higher. The top five hub genes were GABRG2, CACNA1A, CNTNAP2, NOS1, and SCN4B. GABRG2, CNTNAP2, and SCN4B in the hub genes have high diagnostic value in determining whether hypopharyngeal carcinoma patients have combined esophageal carcinoma (AUC: 0.944, 0.944, 0.972). These genes could possibly be used as potential molecular markers for assessing the risk of co-morbidity of hypopharyngeal carcinoma combined with esophageal carcinoma.
Similar content being viewed by others


Introduction
The HPSCC (hypopharyngeal squamous cell carcinoma) is one of the head and neck tumors with high malignancy. Its incidence rate is about 3–5%1 in head and neck malignancies. Research shows that the prognosis of hypopharyngeal carcinoma is poor, with a recurrence median survival period of 6–7 months and a 5-year recurrence-free survival rate of 30–40%2. The Second Primary Malignancy (SPM) is an independent prognostic factor affecting the survival rate of hypopharyngeal carcinoma, significantly increasing the risk of death in patients with nasopharyngeal carcinoma3. The esophagus is the most common site of SPMs in patients with head and neck squamous cell carcinoma, with an incidence rate of up to 30% and a risk of 28.6 times higher than that of the normal population4.
Hypopharyngeal-esophageal carcinomas have no obvious symptoms at the early stage of the disease, because of the special anatomical position of the region, electronic gastroscopy often passes through this region quickly to reduce the pain of the patients, and laryngoscopy is often not able to peep into the esophagus, so it is difficult to diagnose in the early stage, and the diagnosis is often progressed to the middle and late stages when confirmed. According to the recommendations of the guidelines for the diagnosis and treatment of multiple primary cancers of the hypopharynx-esophagus4, for patients with synchronous hypopharyngeal carcinoma combined with esophageal carcinoma, the comprehensive treatment model of surgical treatment followed by postoperative chemoradiotherapy is an ideal treatment method. However, the complications of surgical treatment, such as gastric emptying dysfunction, gastroesophageal anastomotic leakage, anastomotic stenosis, or pleural effusion, seriously affect the quality of life of patients5. For patients with metachronous hypopharyngeal carcinoma combined with esophageal carcinoma, the treatment options for secondary carcinoma are always limited by thetreatment of the first primary carcinoma, resulting in long treatment cycles andpoor efficacy for patients. Currently, there are no clear reports on the positive efficacy of targeted therapy and immunotherapy for multiple primary carcinomain combination. Gene sequencing is a method for analyzing genome sequences, enabling genetic evolution analysis and prediction of important trait candidate genes, as well as analyzing the base sequence of specific DNA fragments. Identifying hub co-disease genes and their possible regulatory molecular mechanisms in hypopharyngeal carcinoma, and esophageal carcinoma, patients at the molecular level through gene sequencing and bioinformatics analysis is of great significance for early diagnosis, targeted drug development, and individualized treatment of patients with hypopharyngeal carcinoma combined with esophageal carcinoma.
Methods
-
1.
Experimental materials
This study included six cases of hypopharyngeal carcinoma combined with esophageal carcinoma in patients and six patients with hypopharyngeal carcinoma. Postoperative pathology confirmed squamous cell carcinoma of the hypopharynx and esophagus. The age range of all patients was 18–80 years old, and their clinical data were complete. All patients had signed relevant informed consent before surgery. Patients with previous primary tumor history other than hypopharyngeal and esophageal carcinoma, or previousesophageal or hypopharyngeal surgery, radiotherapy, or chemotherapy were excluded. Before taking specimens, the surgeon and pathologist communicated with each other about the sampling site and size of the specimen. Pathological specimens were taken from the hypopharyngeal cancer section. The central part of the tumor without necrosis and infection was selected for tissue specimen collection, and it was ensured that the tumor after being taken would not affect the patient's pathological diagnosis. During the operation, after the specimen was removed from the body, it was rinsed twice with saline. Immediately cut two pieces of non-necrotic tumor tissue with a size of 0.5 cm3, and placed them in EP tubes. Within 30 min, transfer them to the corresponding position of the – 80 ℃ refrigerator for preservation. The pathological diagnosis of the tissue was made by two experienced chief pathologists from the Department of Pathology of Chaoyang Central Hospital by the WHO diagnostic criteria, and it was ensured that the tumor cell ratio of the tumor samples in this study was greater than 80%. The patient’s TNM staging and clinical staging standards were based on the 8th edition of the American Joint Committee on Carcinoma (AJCC) TNM Staging Manual in 2017. This study was approved by the Ethics Committee of Chaoyang Hospital with ethical approval number [2022] 30, and all patients provided informed written consent. Patients clinical information sheet in Table 1.
-
2.
RNA sequencing
Detect the purity of the sample and the concentration and integrity of the RNA sample. Take 1–3 μg total RNA from each sample as the starting material to construct a transcriptome sequencing library. According to the instructions of the library preparation kit, select different index tags to construct the library and use Qubit 3.0 for preliminary quantification. Dilute the library to 1 ng/ul, detect the insert size of the library, and use the Illumina platform for sequencing. Run the PE150 sequencing strategy to obtain 150 bp paired-end sequencing reads. The Raw Data is stored in FASTQ file format, removing reads with adapters, uncertain base information, and low-quality reads (Qphred <= 20 base number accounting for more than 50% of the length of the entire read).
-
3.
Bioinformatics analysis of differential genes
The GO (Gene Ontology) database is an ontology widely used in the field of bioinformatics. It covers three aspects of biology: cellular components, molecular function, and biological processes. The functional enrichment analysis method annotates the selected genes based on the GO database to obtain all the functions involved in the genes and then uses Fisher’s exact test and multiple comparison tests to calculate the significance level and false discovery rate (FDR) for each function. This allows the screening of significant functions embodied by genes, with the criterion of significance screening being p < 0.05.
KEGG (Kyoto Encyclopedia of Genes and Genomes) is a database that integrates genomic, chemical, and system function information. It is a database for systematically analyzing the relationships between genes (and their encoded products), gene functions, and genomic information. It helps researchers study genes and expression information as a whole network. Significance screening criteria: p < 0.05.
Map the differentially expressed gene set to the STRING database (STRING: functional protein association networks, http://cn.string-db.org) to construct a protein interaction network of differentially expressed genes. Import the differentially expressed genes into the STRING database for analysis, and import the output file into Cytoscape for visualization. Using the cytoHubba plugin in Cytoscape, calculate the hub genes based on degree.
-
4.
Use R software packages such as ggplot2, Survival, timeROC, limma, etc. for analysis. Import the hub genes into relevant files and use the aforementioned packages to calculate the ROC curve and nomogram of the hub genes.
-
5.
PCR Verification of gene selection
The six differentially expressed genes with significant differences and relatively high expression levels in the 12 groups of samples were selected for PCR detection: LINC00470, CYP2B7P, NKX2-8, CADM3, MIPOL1 and ZFP82. Gene sequencing and PCR-related reagents are shown in Table 2. Gene and primer information of PCR in Tables 3 and 4. Approximately 100 mg of tumor tissue from the cryopreservation tube was cut with sterile scissors and placed in an RNase-free centrifuge tube. Two steel balls were added, followed by 1000 μL of tissue lysate (Trizol). The tube was centrifuged in a high-channel tissue homogenizer, and the total RNA was extracted using the centrifugation solution and reverse transcribed. After diluting the reverse transcribed cDNA, real-time quantitative PCR was performed using SYBR Green I for gene expression analysis. The relative expression was calculated using the 2−△△Ct method. GraphPad Prism 9.0 statistical software was used to process the data for graphing, and the independent samples t-test was used for measures that conformed to normal distribution, and the nonparametric test was used for measures that were not normally distributed, with p < 0.05 being statistically significant.
Ethical approval
The study was approved by the Ethics Committee of Chaoyang Downtown Hospital and informed written consent was obtained from all patients. Primary tumors and adjacent normal tissues of HPSCC patients were obtained according to the Declaration of Helsinki.
Results
-
1.
Results of differentially expressed genes: A total of 44,876 genes were screened and 76 genes showed significant statistical differences between the hypopharyngeal carcinoma with esophageal carcinomagroup, and the hypopharyngeal carcinoma group (Screening Principle|log2(FoldChange)|> 1 p < 0.05). Among them, 47 genes were expressed lower in the hypopharyngeal carcinoma with the esophageal carcinoma group than in the hypopharyngeal carcinoma group, and 29 genes were expressed higher (Table 5). The sequencing results are shown in Table 6. The results of the differential gene visualization volcano map and heatmap are shown in Figs. 1 and 2. Group A is hypopharyngeal cancer combined with esophageal cancer, Group B is hypopharyngeal cancer.
Table 5 Results for 76 DEGs. Table 6 Sequencing results. Figure 1 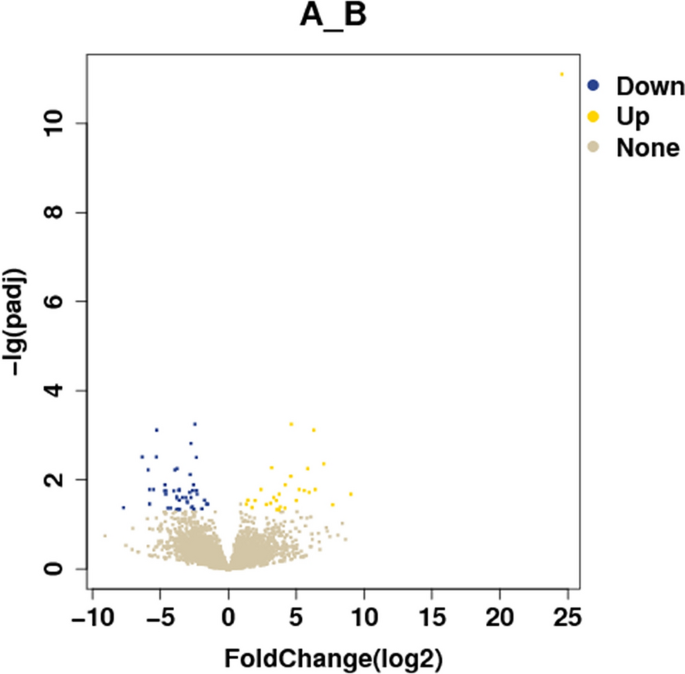
Differential expression genes volcano diagram.
Figure 2 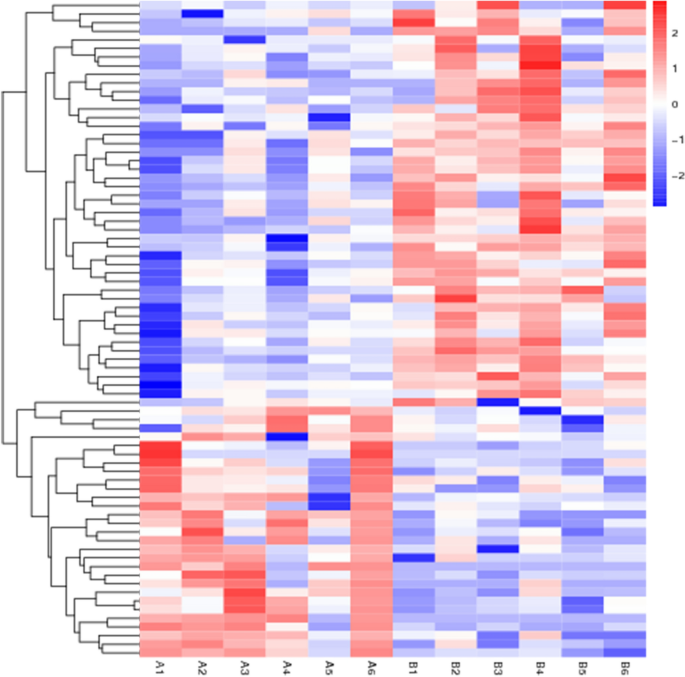
Differential expression genes heatmap.
-
2.
GO analysis results: In GO data analysis, we found that 76 differentially expressed proteins were significantly enriched in 275 GO terms (p < 0.05), including 45 terms related to cellular components (GO-CC). Differentially expressed proteins mainly participate in the formation of cell components such as the transporter complex transmembrane transporter complex, cation channel complex, ion channel complex etal. There are 190 items related to biological processes (GO-BP), such as calcium channel activity, cation channel activity, voltage − gated channel activity, transporter activity et al. And 40 related to molecular function (GO-MF), include gamma − aminobutyric acid signaling pathway, calcium ion transmembrane transport, cell adhesion molecules et al. The GO results are shown in Table 7 and Fig. 3.
Table 7 GO results. Figure 3 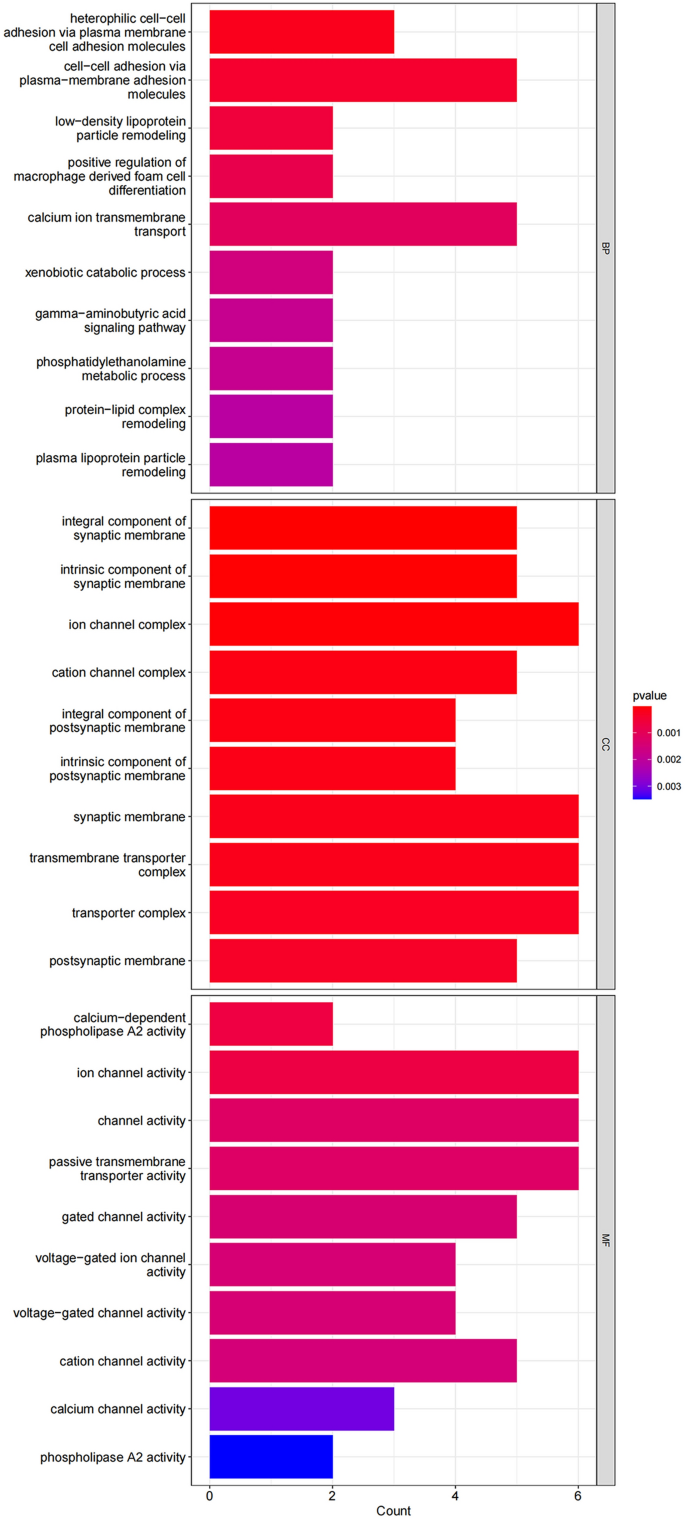
GO result bar chart, showing the top 30 most significant GO items for BP, CC, and MF.
-
3.
KEGG analysis results: KEGG analysis was performed on 76 differentially expressed genes, and KEGG enrichment results were screened using a p < 0.05. A total of 29 signaling pathways were identified. The results showed that the differentially expressed genes were mainly involved in carcinoma-related pathways, including cell adhesion molecules, GABAergic synapses, calcium signaling pathways, α-linolenic acid metabolism, and linoleic acid metabolism. The KEGG results are shown in Table 8 and Fig. 4.
Table 8 KEGG results. Figure 4 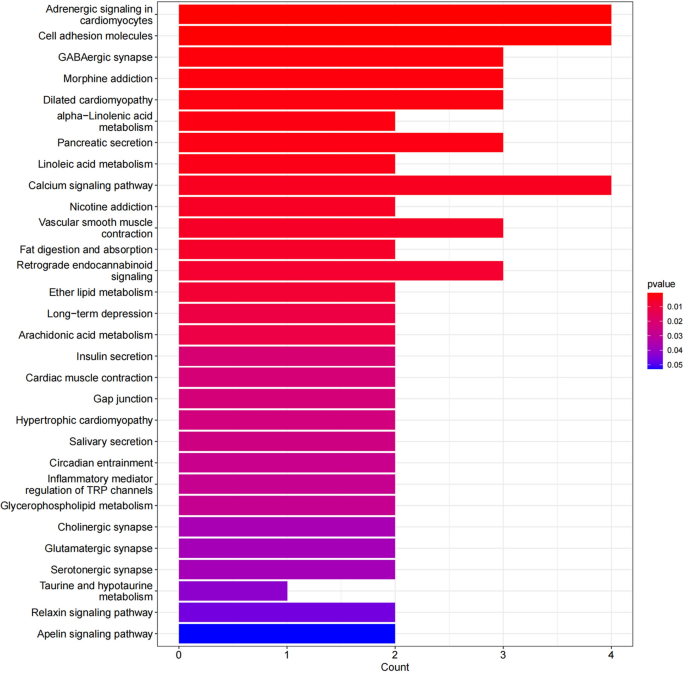
Significantly enriched KEGG pathways of DEGs in HPSCC.
-
4.
PPI analysis: After importing the differentially expressed genes into the STRING database (https://string-db.org/), the output file was imported into Cytoscape software for visualization. The cytoHubba plug-in in Cytoscape was used to calculate the hub genes based on degree. The top five genes were GABRG2, CACNA1A, CNTNAP2, NOS1, and SCN4B, with degrees of 10, 9, 8, 7, and 7. The PPI results are shown in Figs. 5, 6 and 7.
Figure 5 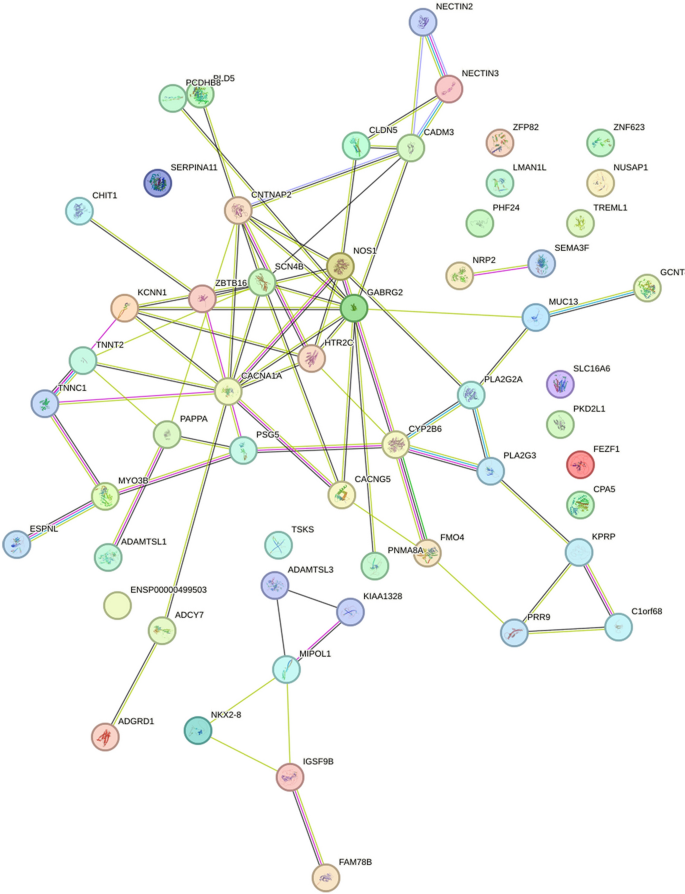
The visualization results of differentially expressed genes in the STRING database.
Figure 6 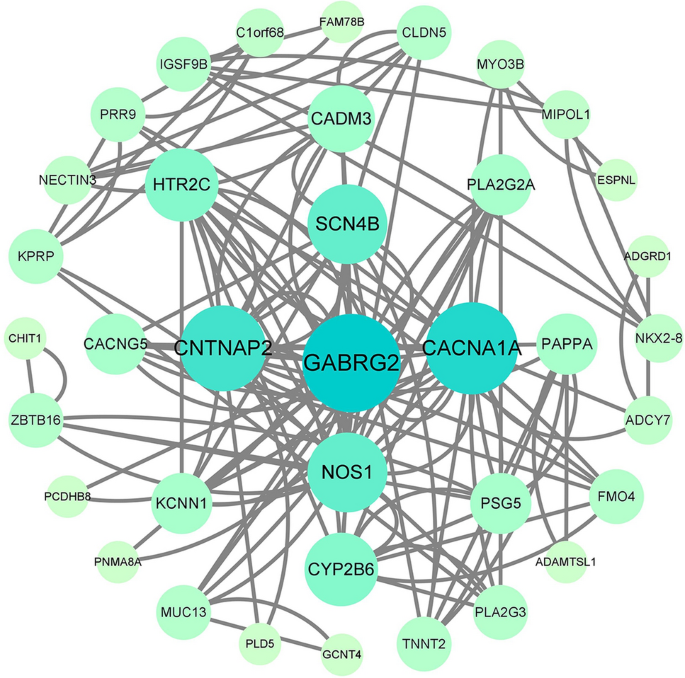
The protein interaction network results in differentially expressed genes, where nodes represent genes and lines represent interactions between two genes. The more lines connected to a node, the greater its connectivity, indicating the importance of the node gene in the network.
Figure 7 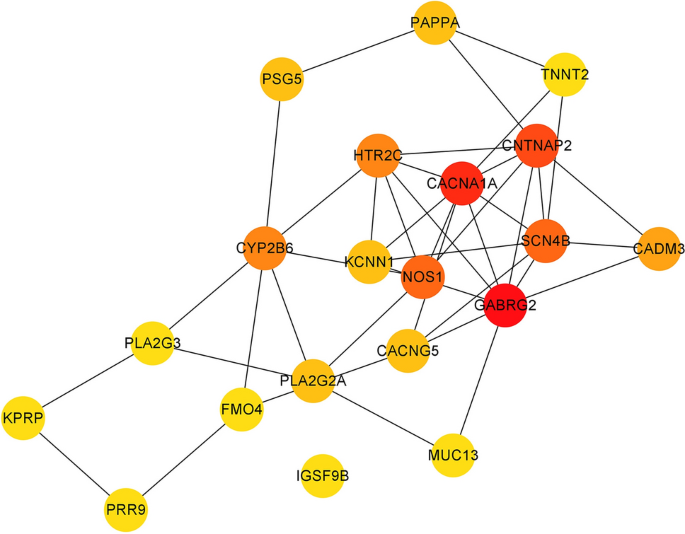
The visualization results of hub genes.
-
5.
Evaluation of hub genes in clinical diagnosis and treatment
The diagnostic value of these hub genes was evaluated using ROC analysis. The ROC curve is a plot of the true positive rate (sensitivity) against the false positive rate (1-specificity) based on a series of different binary classifications (cut-off values or decision thresholds). The area under the curve (AUC) is used to determine the accuracy of diagnosis. In principle, the diagnostic results do not match the actual situation when the AUC < 0.5, while AUC = 0.5 indicates no effect at all. When 0.5 < AUC < 0.7, 0.7 < AUC < 0.9, and AUC > 0.9, the diagnostic effect is low, medium, and high, respectively, suggesting that GABRG2, CNTNAP2, and SCN4B have high diagnostic value for patients with hypopharyngeal carcinoma combined with esophageal carcinoma, while NOS1 and CACNA1A have moderate diagnostic value. The ROC and nomogram results are shown in Figs. 8 and 9.
Figure 8 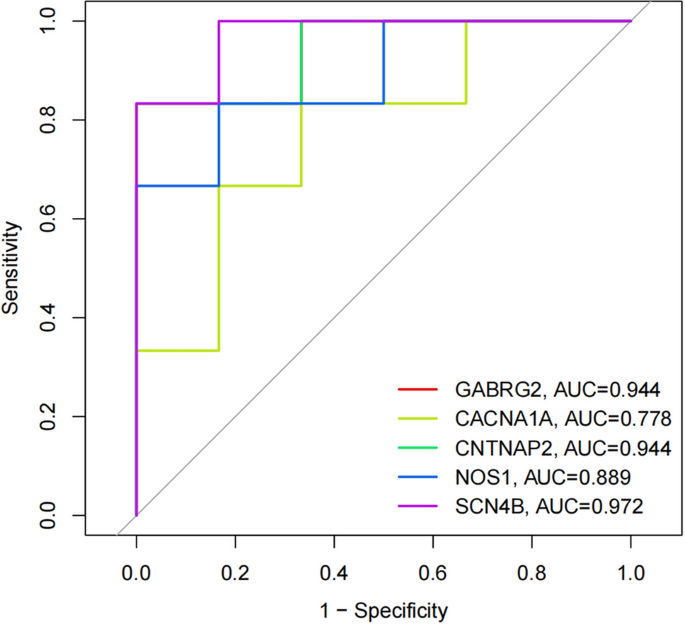
The ROC curve results for the hub genes.
Figure 9 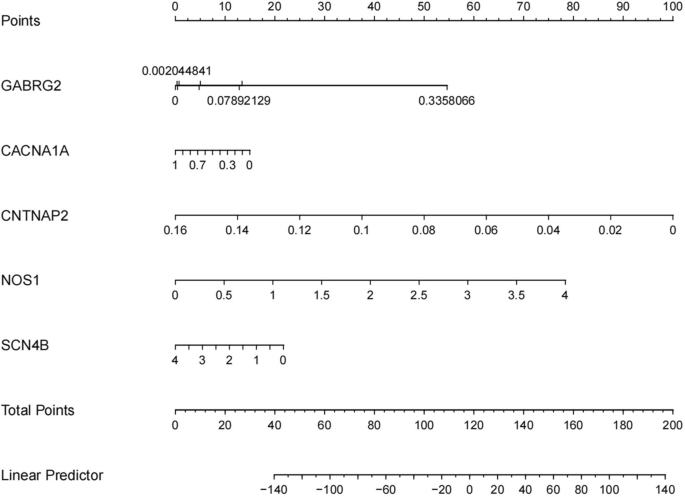
The nomogram for the hub genes. Based on the results of patient genetic testing, corresponding shubs can be obtained for the expression level of each gene. Adding all score hubs can predict the risk of comorbidity of esophageal carcinoma in patients with hypopharyngeal carcinoma.
-
6.
The results of PCR to detect the expression of the six differentially expressed genes with significant differences and relatively high expression levels—LINC00470, CYP2B7P, NKX2-8, CADM3, MIPOL1 and ZFP82 are shown in Fig. 10,* indicates p < 0.05, *** indicates p < 0.001, and the results are statistically significant, and the detection of the expression is basically the same as the sequencing results. Group A is hypopharyngeal cancer combined with esophageal cancer, Group B is hypopharyngeal cancer.
Figure 10 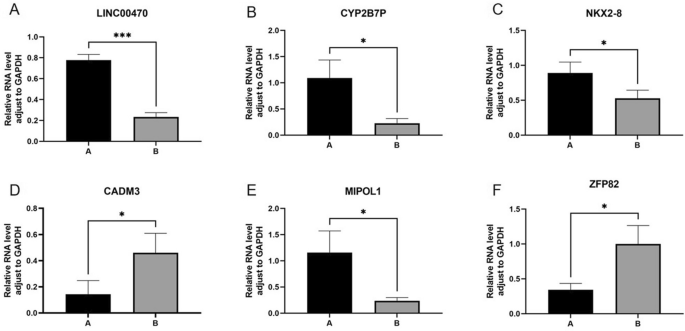
The results of PCR detection of LINC00470, CYP2B7P, NKX2-8, CADM3, MIPOL1 and ZFP82 are shown in Fig. 9, * indicates p < 0.05, *** indicates p < 0.001, and the results are statistically significant, and the detection of the expression is basically the same as the sequencing results. Group A is hypopharyngeal cancer combined with esophageal cancer, Group B is hypopharyngeal cancer.










Discussion
The theoretical basis for the occurrence and development of hypopharyngeal carcinoma combined with esophageal carcinoma is widely supported by the field cancerization theory first proposed by Slaughter et al. in 1953 during the analysis of oral squamous cell carcinoma6. However, the current mechanism of the occurrence and development of hypopharyngeal carcinoma combined with esophageal carcinoma is not well understood, and various studies have suggested that there may be co-morbid factors in hypopharyngeal carcinoma combined with esophageal carcinoma. Studies have shown that when PD-L1 is used as a predictive biomarker for immunotherapy for hypopharyngeal carcinoma combined with esophageal carcinoma, the immune-related expression of tumor tissues in patients with hypopharyngeal carcinoma combined with esophageal carcinoma is significantly correlated based on PCR and immunohistochemical results7. There was a study conducted bioinformatics analysis on independent datasets of hypopharyngeal carcinoma combined with esophageal carcinoma in the GEO database and identified miR-29 as the most frequently interacting miRNA with hub genes8. However, the conclusion still needs to be verified by clinically relevant experiments. Another research evaluated the expression of proteins related to hypopharyngeal carcinoma combined with esophageal carcinoma and detected the overexpression of p53 and the deletion of Fhit expression in the relevant tissues by immunohistochemistry9. The above studies suggest that there may be common pathogenic genes between hypopharyngeal carcinoma combined with esophageal carcinoma.
In this experiment, we screened the differential genes between hypopharyngeal carcinoma combined with esophageal carcinoma and hypopharyngeal carcinoma based on RNA-seq and verified the results by PCR, which were basically consistent with the results of the gene screening, and then assessed the utility of the hub genes screened by bioinformatics and constructed the prediction model for their utility in clinical diagnosis and treatment. Among them, GABRG2 and CACNA1A were up-regulated, while genes CNTNAP2, NOS1 and SCN4B were down-regulated. These five genes are all related to the occurrence and development of tumors and can be used as hub genes for predicting the risk of patients with hypopharyngeal carcinoma combined with esophageal carcinoma. To the best of our knowledge, there have been no previous studies reporting the association between GABRG2, CACNA1A, CNTNAP2, NOS1, and SCN4B and hypopharyngeal carcinoma combined with esophageal carcinoma. Therefore, these genes may represent new biomarkers or may exhibit gene fusion in hypopharyngeal carcinoma combined with esophageal carcinoma. However, their specific mechanisms in the occurrence and development of hypopharyngeal carcinoma combined with esophageal carcinoma require deeper exploration.
GABA is the most abundant inhibitory neurotransmitter, and its receptor is an important pharmacological target for many antiepileptic drugs.GABRG2 is located on chromosome 5 and encodes the γ2 subunit of GABAAR, which plays an important role in receptor trafficking and post-synaptic membrane aggregation10. It is the most common epileptic pathogenic gene among GABAAR subunits11. It is abnormally expressed in various cancers such as primary glioblastoma, hepatocellular carcinoma, and colon adenocarcinoma12,13,14. There is relatively little research on GABRG2 in head and neck squamous cell carcinoma. Related studies have found that GABRG2 may regulate laryngeal cancer recurrence by participating in the EMX2OS-miR-124-
CALCA/GABRG2 axis15, but the conclusions of this study need to be further verified by relevant molecular experiments. As the gene with the strongest degree of interaction in PPI, the biological role of GABRG2 overexpression in hypopharyngeal carcinoma with esophageal carcinoma deserves further study through in vitro and in vivo research.
CNTNAP2 is located on chromosome 7q35 and encodes a presynaptic type I transmembrane protein CASPR2, which is involved in cell–cell adhesion and synaptic interactions in the nervous system16.CNTNAP2 is associated with various neurodevelopmental disorders such as tic-toilet syndrome, intellectual disability, autism spectrum disorder (ASD), and schizophrenia17. In aggressive oligodendroglioma, CNTNAP2 expression decreases and is associated with reduced overall survival18. In a study of head and neck squamous cell carcinoma, CNTNAP2 has some predictive ability for the sensitivity of laryngeal squamous cell carcinoma to induction chemotherapy19.
VGSC (Voltage-gated sodium channel) is a key protein that transmits molecular information.SCN4B (Sodium Voltage-Gated Channel Beta Subunit 4 ) is located on chromosome 11q23.3 and encodes a protein consisting of 228 amino acid residues20. SCN4B mutations are associated with a variety of diseases, including cancer, epilepsy, arrhythmia, sudden infant death syndrome, neuropathic pain, and various neurodegenerative diseases21. As a tumor suppressor gene, SCN4B plays a role in various cancers. Emeline22 and others found that SCN4B expression was downregulated in breast cancer cells and promoted the migration and invasion of breast cancer cells through in vivo and in vitro experimental studies. The expression of SCN4B in the tissues of thyroid cancer patients is down-regulated, and SCN4B expression is an independent predictor of good relapse-free survival in thyroid cancer patients23. In prostate cancer cells, miR-3175 expression increases, and SCN4B expression decreases. However, after knocking down miR-3175, cell growth, migration, invasion, and N-cadherin expression, which are related phenotypes, are inhibited, and SCN4B expression is upregulated, suggesting that SCN4B may be a potential mechanism for miR-3175 to promote prostate cancer cell growth and invasion24.
Nitric oxide is a signaling molecule synthesized by three subtypes of NO synthase (NOS1, NOS2, and NOS3) and is increased in various cancers and involved in various cancer processes, such as proliferation and migration25. NO has a dual effect on tumors. When present at low-moderate concentrations, NO can stimulate various carcinogenic signaling pathways, such as AKT, ERK, and HIF. When present at high concentrations, NO can produce nitrosative stress and stimulate anti-cancer signaling mechanisms, such as p53 and apoptosis pathways26. Gesche et al. evaluated relevant tissue samples from 30 patients with primary oropharyngeal squamous cell carcinoma using immunohistochemistry and RT-PCR and found that NOS1 and NOS3 expression significantly increased27. NOS1 can participate in the development of various cancers through protein S-nitrosylation, which is a covalent post-translational modification that results in the coupling of NO moieties containing active thiol groups with protein cysteine residues to form S-nitrosothiol (SNO) moieties28. NOS1 reduces the recruitment of STAT1-mediated HDAC2 to ISG promoters and promotes lung metastasis of melanoma through the S-nitrosylation of HDAC4-C16/C229. NOS1 can also activate the AKT/mTOR signaling pathway through the S-nitrosylation of PTEN, thereby inhibiting autophagy in nasopharyngeal carcinoma cells30.
CACNA1A (Calcium Voltage-Gated Channel Subunit Alpha1 A) is located on the short arm of chromosome 19 at position 13.13 and encodes the channel-forming protein alpha-1A subunit of the P/Q-type voltage-gated calcium channel31. This channel mediates Ca2+ ion entry into excitable cells and participates in various calcium-dependent biological processes such as muscle contraction, neurotransmitter, and hormone release. CACNA1A mutations are associated with various neurological diseases such as migraine, epilepsy, paroxysmal ataxia, and spinocerebellar ataxia32. In primary glioblastoma, CACNA1A expression is upregulated and involved in the development of cancer stem cells (CSC) and the regulation of related signaling pathways33. When studying the effect of radiotherapy in patients with oropharyngeal squamous cell carcinoma, Tomoya Kurokawa et al. found that a gene set consisting of eight genes, including CACNA1A, could be used for prediction and had high predictive power34.
Combining previous research on five hub genes, we found that the research on detecting the expression of hub genes was basically consistent with our results, and the PCR results also verified the accuracy of our sequencing results. The mechanism of promoting the occurrence of various tumors also suggests that the hub genes are multidimensional in the process of the occurrence and development of hypopharyngeal carcinoma combined with esophageal carcinoma. According to the results of GO and KEGG, the hub genes mainly constitute cell membrane channel proteins such as ion channel complexes, transmembrane transporter complexes, and transporter complexes, and participate in the transmembrane transport and signal transduction of related substances. Ion channels are responsible for the flow of ions across the cell membrane and play a very important role in human physiology, including the regulation of ion homeostasis, electrical excitability, and cell signaling. However, when the expression of ion channels changes, these channels can cause various channel diseases, including cancer. Specific types of ion channels can participate in different stages of tumor progression and mediate the expression of multiple phenotypes of tumors, such as increasing cell heterogeneity by selectively expressing malignant cell clones that support proliferation, migration, or invasion through the selective expression of ion channel types35. In head and neck squamous cell carcinoma, voltage-gated K+ channels are abnormally expressed and serve as specific markers in the early stages of tumorigenesis and the late stages of disease progression36. Studies have shown that the incidence rate of laryngeal cancer in patients with positive dysplasia of Kv3.4 K+ channel subunits is significantly increased37. When the expression of Kv3.4 is inhibited, it can selectively block the cell cycle in the G2/M phase and then inhibit cell proliferation. In esophageal squamous cell carcinoma, overexpression of the voltage-gated potassium channel subfamily KCNJ15 can lead to increased cell proliferation38. These studies suggest that one of the mechanisms underlying the development and progression of hypopharyngeal carcinoma with co-occurring esophageal carcinoma may be the overexpression or suppression of genes that mediate ion channel-related proteins, leading to abnormal protein function or altered expression. In addition to K+ ion channels, Ca2+, Na+, and Cl- channels have also been associated with cancer. The biological role of the aberrant expression of ion channel-associated proteins mediated by hub gene in hypopharyngeal carcinoma combined with oesophageal carcinoma deserves to be further investigated by in vitro and in vivo experiments, and the relevant contents include the upstream and downstream regulatory genes and related protein expression of hub gene, the pathway of regulation and the related biological functions. This will be our research direction in the next stage.
The onset of hypopharyngeal carcinoma is insidious, and it often progresses to the middle and late stages when diagnosed, with poor prognosis. When combined with a second primary cancer, it can seriously affect the patient's prognosis and survival. Currently, the main method for determining whether hypopharyngeal carcinoma is associated with a second primary cancer of the esophagus is NBI staining endoscopy combined with biopsy. The procedure often causes patients to experience significant pain, and due to the specific anatomical ___location of the hypopharynx and esophagus, there is a risk of human error during the operation, resulting in missed detection. Therefore, from an objective perspective, molecular markers related to gene detection in patients' blood or other secretions will provide better-individualized treatment options for early detection, diagnosis, and prediction of hypopharyngeal carcinoma patients with or without esophageal carcinoma. This study screened the differentially expressed genes between hypopharyngeal carcinoma with and without esophageal carcinoma by gene sequencing and validated the sequencing results through QRT-PCR experiments. We further used bioinformatics analysis to identify the hub genes of hypopharyngeal carcinoma with or without esophageal carcinoma, suggesting that GABRG2, CACNA1A, CNTNAP2, NOS1, and SCN4B genes may be potential molecular markers for indicating the risk of hypopharyngeal carcinoma co-occurring with esophageal carcinoma. This study will improve the detection rate of precancerous lesions in the esophageal region, which is beneficial for the early diagnosis and treatment of hypopharyngeal carcinoma with or without esophageal carcinoma, as well as the monitoring and follow-up of hypopharyngeal carcinoma after treatment. However, there are still shortcomings in this experiment, such as the small sample size selected, the lack of basic research on the specific mechanisms of hub genes in the occurrence of hypopharyngeal carcinoma with or without esophageal carcinoma, and the potential differences in genes between patients from different regions and ethnic groups. It is unclear whether the diagnostic value of hub genes for hypopharyngeal carcinoma with or without esophageal carcinoma is consistent with the actual clinical situation. Therefore, we will adopt a multi-center study to supplement the number of cases to obtain more data, so that our conclusions are more accurate, to exclude regional and ethnic differences. In the follow-up study, we will add genomic analysis and comparison between normal mucosal tissue and oesophageal lesions to validate the results of our current study and make our conclusions more scientific and accurate.We will further improve cell and animal experiments to explore the mechanisms of hub genes in the occurrence of hypopharyngeal carcinoma with or without esophageal carcinoma. We will adopt more detailed follow-up strategies to fully understand the changes in the condition of hypopharyngeal carcinoma patients. We hope to further improve and refine these problems and deficiencies through subsequent experiments.
Data availability
The original contributions presented in the study are included in the article Material. Further inquiries can be directed to the corresponding authors.
Abbreviations
- HPSCC:
-
Hypopharyngeal squamous cell carcinoma
- SPM:
-
Second primary malignancies
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PPI:
-
Protein–Protein interaction
- BP:
-
Biological processes
- CC:
-
Cellular components
- MF:
-
Molecular functions
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- AUC:
-
Area under curve
- GABA:
-
γ-Aminobutyric acid
- GABRG2:
-
Gamma-Aminobutyric acid type A receptor subunit Gamma2
- GABAAR:
-
γ-Aminobutyric acid type A receptor
- CNTNAP2:
-
Contactin Associated Protein 2
- VGSC:
-
Voltage-gated sodium channel
- SCN4B:
-
Sodium voltage-gated channel beta subunit 4
- CACNA1A:
-
Calcium voltage-gated channel subunit Alpha1 A
References
Kawakita, D. et al. Trends in the incidence of head and neck cancer by subsite between 1993 and 2015 in Japan. Cancer Med. 11(6), 1553–1560 (2022).
Visini, M., Giger, R., Shelan, M., Elicin, O. & Anschuetz, L. Predicting factors for oncological and functional outcome in hypopharyngeal carcinoma. Laryngoscope 131(5), E1543–E1549 (2021).
Lee, D. H. et al. Second cancer incidence, risk factor, and specific mortality in head and neck squamous cell carcinoma. Otolaryngol. Head Neck Surg. 149(4), 579–586 (2013).
Committee of esophageal carcinoma in China Anti-Cancer A, Chinese Working Group on Cooperative D, Treatment of H, Esophageal C. Chinese expert consensus on multiple primary cancers of hypopharynx and esophagus. Zhonghua Wai Ke Za Zhi. 58(8), 589–595 (2020).
Thompson, C. S. G. et al. Complications and predisposing factors from a decade of total laryngectomy. J. Laryngol. Otol. 134(3), 256–262 (2020).
Slaughter, D. P., Southwick, H. W. & Smejkal, W. Field cancerization in oral stratified squamous epithelium; Clinical implications of multicentric origin. Cancer. 6(5), 963–968 (1953).
Chen, T. C. et al. The differences of immunologic and TP53 mutant phenotypes between synchronous and metachronous head and neck cancer and esophageal carcinoma. Oral Oncol. 111, 104945 (2020).
Zhou, R., Liu, D., Zhu, J. & Zhang, T. Common gene signatures and key pathways in hypopharyngeal and esophageal squamous cell carcinoma: Evidence from bioinformatic analysis. Med. (Baltim.). 99(42), e22434 (2020).
Yamamoto, S. et al. Frequent aberrant p53 and Fhit expression in endoscopically resected superficial hypopharyngeal carcinoma combined with esophageal carcinoma. Oncol. Lett. 14(1), 587–592 (2017).
Kerti-Szigeti, K., Nusser, Z. & Eyre, M. D. Synaptic GABAA receptor clustering without the gamma2 subunit. J. Neurosci. 34(31), 10219–10233 (2014).
Kang, J. Q. & Macdonald, R. L. Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to Dravet syndrome. JAMA Neurol. 73(8), 1009–1016 (2016).
Yan, L. et al. Distinct diagnostic and prognostic values of gamma-aminobutyric acid type A receptor family genes in patients with colon adenocarcinoma. Oncol. Lett. 20(1), 275–291 (2020).
Zhang, Y. A. et al. RNA binding protein Nova1 promotes tumor growth in vivo and its potential mechanism as an oncogene may due to its interaction with GABA(A) Receptor-gamma2. J. Biomed. Sci. 23(1), 71 (2016).
Gonzalez-Garcia, N. et al. Multivariate analysis reveals differentially expressed genes among distinct subtypes of diffuse astrocytic gliomas: diagnostic implications. Sci. Rep. 10(1), 11270 (2020).
Tang, Z., Wei, G., Zhang, L. & Xu, Z. Signature microRNAs and long noncoding RNAs in laryngeal cancer recurrence identified using a competing endogenous RNA network. Mol. Med. Rep. 19(6), 4806–4818 (2019).
D’Onofrio, G. et al. Genotype-phenotype correlation in contactin-associated protein-like 2 (CNTNAP-2) developmental disorder. Hum. Genet. 142(7), 909–925 (2023).
Penagarikano, O. et al. The absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and hub autism-related deficits. Cell. 147(1), 235–246 (2011).
Rautajoki, K. J. et al. PTPRD and CNTNAP2 as markers of tumor aggressiveness in oligodendrogliomas. Sci. Rep. 12(1), 14083 (2022).
Li, L. et al. The identification of induction chemo-sensitivity genes of laryngeal squamous cell carcinoma and their clinical utilization. Eur. Arch. Otorhinolaryngol. 275(11), 2773–2781 (2018).
Wisedchaisri, G. et al. Resting-state structure and gating mechanism of a voltage-gated sodium channel. Cell. 178(4), 993–1003 (2019).
Bouza, A. A. & Isom, L. L. Voltage-gated sodium channel beta subunits and their related diseases. Handb. Exp. Pharmacol. 246, 423–450 (2018).
Bon, E. et al. SCN4B acts as a metastasis-suppressor gene preventing hyperactivation of cell migration in breast cancer. Nat. Commun. 7, 13648 (2016).
Gong, Y. et al. Preserved SCN4B expression is an independent indicator of favorable recurrence-free survival in classical papillary thyroid cancer. PLoS ONE. 13(5), e0197007 (2018).
Huang, H., Qing, X. Y., Zhou, Q., Li, H. D. & Hu, Z. Y. Silencing of microRNA-3175 represses cell proliferation and invasion in prostate cancer by targeting the potential tumor-suppressor SCN4B. Kaohsiung J. Med. Sci. 37(1), 20–26 (2021).
Khan, F. H. et al. The role of nitric oxide in cancer: master regulator or NOt?. Int. J. Mol. Sci. 21(24), 9393 (2020).
Thomas, D. D. et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic. Biol. Med. 45(1), 18–31 (2008).
Frohwitter, G. et al. Oxidative and nitrosative stress in oral squamous cell carcinoma. Cells Tissue Organs. 209(2–3), 120–127 (2020).
Zhang, Y., Deng, Y., Yang, X., Xue, H. & Lang, Y. The relationship between protein S-nitrosylation and human diseases: A review. Neurochem. Res. 45(12), 2815–2827 (2020).
Xu, P. et al. NOS1 inhibits the interferon response of cancer cells by S-nitrosylation of HDAC2. J. Exp. Clin. Cancer Res. 38(1), 483 (2019).
Zhu, L. et al. Publisher correction: NOS1 S-nitrosylates PTEN and inhibits autophagy in nasopharyngeal carcinoma cells. Cell Death Dis. 10(4), 286 (2019).
Heck, J. et al. More than a pore: How voltage-gated calcium channels act on different levels of neuronal communication regulation. Channels (Austin). 15(1), 322–338 (2021).
Manickam, A. H. & Ramasamy, S. Mutations in the voltage-dependent calcium channel CACNA1A (P/Q type alpha 1A subunit) causing neurological disorders—an overview. Neurol. India 69(4), 808–816 (2021).
Hsu, J. B. et al. Identification of differentially expressed genes in different glioblastoma regions and their association with cancer stem cell development and temozolomide response. J. Pers. Med. 11(11), 1047 (2021).
Kurokawa, T. et al. Establishment of epigenetic markers to predict irradiation efficacy against oropharyngeal cancer. Cancer Sci. 111(4), 1407–1416 (2020).
Becchetti, A. Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. Am. J. Physiol. Cell Physiol. 301(2). C255–65. https://doi.org/10.1152/ajpcell.00047.2011. Epub 2011 Mar 23. PMID: 21430288 (2011).
Del-Río-Ibisate, N., Granda-Díaz, R., Rodrigo, J. P., Menéndez, S. T., García-Pedrero, J. M. Ion channel dysregulation in head and neck cancers: perspectives for clinical application. Rev. Physiol. Biochem. Pharmacol. 181, 375–427. https://doi.org/10.1007/112_2020_38. PMID: 32789787 (2021).
Menéndez, S. T., et al. Expression and clinical significance of the Kv3.4 potassium channel subunit in the development and progression of head and neck squamous cell carcinomas. J. Pathol. 221(4), 402–10. https://doi.org/10.1002/path.2722. PMID: 20593490 (2010).
Nakamura, S, et al. KCNJ15 Expression and malignant behavior of esophageal squamous cell carcinoma. Ann. Surg. Oncol. 27(7), 2559–2568. https://doi.org/10.1245/s10434-019-08189-8. Epub 2020 Feb 12. PMID: 32052303 (2020).
Funding
The Liaoning Natural Science Foundation Program (No2019-ZD-0901).
Author information
Authors and Affiliations
Contributions
(I) Conception and design:L Li; (II) Administrative support: Central Hospital of Chaoyang; (III) Provision of study materials or patients: Central Hospital of Chaoyang; (IV) Collection and assembly of data: J Zhang (V) Data analysis and interpretation: J Zhang(VI) Collated epidemiologic and clinical content on multiple primary cancers of the hypopharynx and hypopharyngeal cancer: Liangyu Zou,MM*1, Fuxian Tan, Hongmin Wang,Zhenlei Wen, Hongmei,Wang(VII) Manuscript writing: All authors; (VIII) Final approval of manuscript: All authors. Lianhe Li designed this study. Jianing Zhang conducted this study and analyzed the data. Jianing Zhang drafted the manuscript. Lianhe Li revised this study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., Zou, L., Tan, F. et al. Screening of co-expressed genes in hypopharyngeal carcinoma with esophageal carcinoma based on RNA sequencing and Clinical Research. Sci Rep 14, 13796 (2024). https://doi.org/10.1038/s41598-024-64162-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-64162-w
