
Abstract
Following glacial retreat after the last ice age, brown bears (Ursus arctos) recolonised Scandinavia. Previous research based on mitochondrial markers suggests that bears recolonised from both the north and the south, with a contact zone in central Scandinavia. More recently, the Scandinavian brown bear was subjected to a strong population decline with only ca. 130 remaining individuals, due to intense human persecution approximately 100 years ago. Here, we analyse 41 ancient, historical, and modern mitochondrial genomes, to examine the number of female lineages involved in the postglacial recolonisation event and temporal changes in the Scandinavian brown bears’ mitochondrial genetic diversity. Our results support the bi-directional recolonisation hypothesis, indicating multiple mitochondrial lineages from clade 1a possibly followed a southern route, while only a single lineage from clade 3a appears to have followed a northern route. Furthermore, we found that the recent bottleneck had a strong impact on the southern subpopulation, resulting in only one remaining haplotype in the contemporary brown bears. For the northern subpopulation, the impact was moderate, and most haplotypes were retained throughout the bottleneck. By exploring the postglacial recolonisation and recent population pressures, our study enhances understanding of how these factors have influenced the genetic diversity of Scandinavian brown bears.
Similar content being viewed by others

Introduction
The flora and fauna of Europe have been profoundly shaped by the last ice age and the following warming climate of the Holocene1. Population contraction into glacial refugia during periods of harsh climate conditions was key for the survival of temperate species1,2,3. Glacial refugia were ice-free areas, mostly found in southern Europe, e.g. Iberia, Italy, the Balkans, and the Caucasus4,5. However, northern refugia have also been described6, such as in the Carpathians7, the Ural mountains8, and Andøya in Norway9. The potential genetic consequences of isolation in refugia have been described in the expansion contraction model2. It states that the isolation in refugia during harsh climate conditions leads to genetic differentiation, while during periods with favourable climate, species expand their ranges, resulting in possible overlap and admixture. At the end of the last ice age, temperate species expanded from isolated refugia into Europe. This postglacial recolonisation has been proposed to be one of the main drivers of the phylogeographic patterns found today1,2.
The Last Glacial Maximum (LGM) was reached between 26.5 and 19 kya (thousand years ago)10, during which northern Europe was covered by the Fennoscandian ice sheet11. After reaching its maximum span around 21 kya, the Fennoscandian ice sheet started to retreat11, which opened the landscape for a recolonising flora and fauna. The deglaciation of Scandinavia started around 16 to 15 kya and Scandinavia became virtually ice free ca. 9.7 kya, when the last remains of the ice sheet in Sarek, Sweden melted12. There are four main hypothesis regarding the postglacial recolonisation of Scandinavia2: (1) A northern route through Finland, observed in species like arctic foxes13, red foxes14, and wood lemmings15; (2) A southern route via land bridges to Sweden, known to be taken by European hedgehogs16; (3) Recolonisation from both directions, seen in common toads17, shrews18, humans19, and field voles20; (4) Expansion from local refugia within Scandinavia, as hypothesised for pine and spruce9,21, and Norwegian lemmings22.
In the context of phylogeography and conservation genetics, the European brown bear (Ursus arctos) represents a key study species. Its current phylogeographic patterns offer valuable insights into the impact and consequences of past climatic changes and human actions23,24,25,26. For many years, brown bears have been considered in a phylogeographic context as a temperate species that primarily survived the last glacial period in the established southern refugia1,3. The results of early phylogeographic studies based on short mitochondrial sequences suggested a postglacial recolonisation of Europe from refugia in Iberia, Italy and the Balkans, and the Caucasus or Carpathian mountains, of bears belonging to mitochondrial western clades 1a, 1b, and eastern clade 3a respectively24,27,28,29. However, Ersmark et al.30 revealed a more complex history of European brown bears during the last ice age. Their findings suggested that brown bears belonging to mitochondrial clade 1a not only survived in the established southern refugia, but also persisted in less commonly recognised mid-latitude European locations throughout the LGM, including areas such as northern France and Belgium.
Previous studies have suggested that brown bears recolonised Scandinavia from two directions1,24,27. Based on phylogeographic studies using short mitochondrial sequences, it has been proposed that brown bears from the Western European refugia recolonised Scandinavia through the southern route over the opening land bridges, while brown bears from the Eastern European refugia expanded into northern Scandinavia via Finland24,25,30,31,32. The bi-directional recolonisation resulted in the presence of two distinct haplogroups, the western (1a) in the South and the eastern (3a) in the North of Scandinavia. Today, the two haplogroups meet within a contact zone in central Scandinavia33, which has been proposed to be a remnant of the bi-directional postglacial recolonisation and likely was retained due to female philopatry in brown bears32. However, Ersmark et al.30 reported early Holocene remains of brown bears in southern Scandinavia carrying a clade 3 haplotype, which could indicate that Eastern clade 3a brown bears might have recolonised Scandinavia through both routes, and that recent processes may have further affected the present-day genetic structure and diversity.
In the early 20th century, the Scandinavian brown bear experienced a dramatic population decline due to intense human persecution34,35. In the mid 1800’s the brown bear population size was ca. 1650 individuals in Sweden and ca. 3100 individuals in Norway, dropping to 130 surviving individuals in Sweden and a virtual extinction in Norway by the 1930’s35. To rescue the species from extinction, protective actions were taken around the early 20th century by the Swedish and later the Norwegian governments34,35, which success was reflected in a subsequent population recovery35,36,37, with an estimate of around 2900 brown bears flourishing in the forests of Sweden by 201738. Today, the brown bear’s distribution in Scandinavia is limited to the northern and central regions, as well as a limited area south of, but close to, the contact zone in Jämtland, Sweden34,38. The mitochondrial diversity of Scandinavian brown bears in the Holocene, and to which extent it has been impacted by the intense population decline, has been addressed in previous studies based on short mitochondrial markers32,39. Bray et al.32 attributed low mitochondrial diversity primarily to the postglacial recolonisation and founder effects, and Xenikoudakis et al.39 emphasised the impact of the recent bottleneck on the mitochondrial diversity loss.
In this study, we present an analysis of 41 mitochondrial genomes of Scandinavian brown bears. We aimed to further elucidate the postglacial recolonisation of Scandinavia, with a focus on achieving a clearer phylogenetic resolution. Moreover, we investigated how recent human impacts, leading to near extinction, have influenced the lineage structure and diversity of Scandinavian brown bears based on mitochondrial data.
Results
Dataset
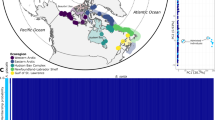
The study focuses on Scandinavian brown bear samples (Ursus arctos) including three ancient specimens dated between 10,290 and 1,916 cal years BP, 17 historical samples from the 1830s to before 1953, and 21 modern samples collected from 2007 to 2012 (Fig. 1). We generated 41 mitogenomes with a minimum average coverage of 5X. The 21 modern samples yielded an endogenous DNA content above 99%, the 17 historical samples had a varying endogenous DNA content between 3 and 90%, and the endogenous DNA content of the three ancient samples was between 0.4 and 11% (Supplementary Tables S2, S3).

The sampling locations of the 41 brown bears (Ursus arctos). Brown bears carrying mitochondrial clade 1a are displayed in red and carrying clade 3a are displayed in blue. Symbols indicate the time period, with dark-coloured circles representing the three ancient samples (early Holocene; marked with star) and the 17 historical pre-bottleneck samples (1830 to before 1953), and light-coloured triangles representing the modern post-bottleneck samples (2007–2012). The dotted line represents the approximate geographic ___location of the contact zone of the two mitochondrial clades. The map was generated using the R package rworldmap40.
Phylogenetic relationship and divergence
We constructed a Bayesian phylogenetic tree of 449 brown bear and polar bear mitogenomes, consisting of the 41 Scandinavian brown bear samples generated in this study and 408 previously published samples (Fig. 2). The mitochondrial clades 1–5 were represented, with the 26 polar bears forming clade 2b. The Bayesian phylogeny supported the presence of brown bears from two distinct mitochondrial clades in Sweden, clade 1a and clade 3a. All Scandinavian brown bears originating from south of the contact zone fall into clade 1a, while all brown bears located north of the contact zone were found within clade 3a. This pattern was consistent for ancient, historical, and modern samples.

Time-calibrated phylogeny of 449 brown and polar bear mitogenomes (15,118 bp) generated in BEAST v1.10.4, using a substitution rate of 2.48 × 10− 8 substitutions per site per year. The y-axis is given in years ago. Individuals are labelled with SampleID or Genbank Accession Number, time period, and country. Collapsed clades are labelled by number. Samples from Scandinavian brown bears sequenced in this study are shown in red (clade 1a) and blue (clade 3a). Branch posterior probability until clade level is displayed, and bars indicate the 95% highest posterior density (HPD) for nodes.
The Scandinavian brown bears assigned to clade 1a clustered together with brown bears from France and Spain, which were previously identified as clade 141,42 (Figs. 2 and 3). The estimated divergence time of the Scandinavian brown bear lineage from its Spanish and French relatives was estimated to 27.1 kya (95% HPD: 34.2–20.8 kya). The time of the most recent common ancestor of all southern Scandinavian brown bears was estimated to 23.7 kya (95% HPD: 30.7–17.5 kya), when the oldest sample (Sw4; 10290 ± 57 calibrated years before present) diverged from the remaining samples. All post-bottleneck southern Scandinavian brown bears cluster together, with a most recent common ancestor estimated to 4.1 kya (95% HPD: 7.3–1.9 kya).

Time-calibrated phylogeny of clade 1a, zoomed in from Fig. 2. The y-axis is given in years ago. Samples from this study are displayed in red. Labelling follows Genbank Accession number or SampleID, timeframe categories, and country. Posterior probability and the 95% HPD of the oldest node is displayed. For the five oldest nodes, within the Scandinavian brown bears, marked with bear symbols, the median node age and 95% HPD in kya are listed in the table.
Clade 3a comprised the northern Scandinavian brown bears and previously published brown bears from Finland, Estonia, Russia, Romania, Bulgaria, Slovakia, Mongolia, USA, Canada, and central Hokkaido (Figs. 2 and 4). The northern Scandinavian brown bears from Norway and Sweden are nested within a clade comprising some brown bears from Finland as well. The estimated divergence time between this group and the remaining clade 3a bears was estimated to 15.9 kya (95% HPD: 19.8–12.2 kya). The most recent common ancestor for this joint Scandinavian and Finnish group was estimated to 11.4 kya (95% HPD: 15.0–8.1 kya).

Time-calibrated phylogeny of clade 3a, zoomed in from Fig. 2. New samples presented in this study are displayed in blue. Labels correspond to SampleID or Genbank accession number, time period, and country, for individual samples and collapsed branches respectively. The node posterior probability is given together with the node branch as 95% HPD for the node forming the clade. The median node ages with 95% HPD in kya of the six oldest nodes within the Scandinavian bears are given in the table, corresponding to the coloured bear symbols. The y-axis is labelled in years ago.
Mitogenome haplotypes and diversity
Consistent with previous studies, we found two distinct haplogroups within the 41 Scandinavian brown bear mitogenomes. All samples from southern Scandinavia fall into the western haplogroup of clade 1a, while all samples from northern Scandinavia carry the eastern haplogroup of clade 3a. Before the bottleneck, a total of 13 haplotypes in 20 brown bears was observed. In the 21 post-bottleneck brown bears, only four haplotypes were found (Fig. 5; Supplementary Figure S2).

Temporal haplotype network of the 41 brown bear mitogenomes (11,412 bp). Red indicates the southern Scandinavian samples, carrying the western haplogroup belonging to clade 1a. Blue represents the northern Scandinavian brown bears, carrying the eastern haplogroup belonging to clade 3a. The number of mutations that separate the two haplogroups are given in square brackets. The network is separated by time, before (bottom) and after the bottleneck (top). Coloured circles display individual haplotypes, labelled with haplotype ID, and are connected by solid lines. Empty circles represent haplotypes that are not found in the corresponding time period and are connected by dashed lines. Black dots on the lines represent missing haplotypes. Haplotypes that are continuous throughout the two time periods are connected by vertical lines. The size of the ellipse indicates the number of individuals assigned to a haplotype. An asterisk next to the haplotype ID indicates the ancient samples.
For the southern Scandinavian brown bears, nine haplotypes among nine pre-bottleneck samples and one haplotype among ten post-bottleneck samples were present. All post-bottleneck southern samples share a haplotype with the pre-bottleneck sample GX0032, from Jämtland. The three ancient southern samples, Sw1, Sw4, and Sw9, carry the haplotypes W1, W2, and W3 respectively, with Sw1 and Sw4 being separated by three and Sw1 and Sw9 being separated by four segregating sites, respectively.
Among the brown bears from northern Scandinavia, the eleven pre-bottleneck samples showed four different haplotypes. Three of those haplotypes were retained in the eleven post-bottleneck northern samples. The majority (19 out of 22) of the northern samples were assigned to haplotype E1 and E2, with six pre- and five post-bottleneck, and three pre- and five post-bottleneck samples, respectively. Haplotype E3 was found only in a single sample, GX0026, and not present in the post-bottleneck data, while haplotype E4 was represented by one sample before and after the bottleneck, GX0061 and IM31.
We observed the highest haplotype and nucleotide diversity among southern Scandinavian brown bears before the bottleneck, and the lowest among the southern Scandinavian brown bears after the bottleneck (Table 1). Haplotype and nucleotide diversity in the post-bottleneck southern samples was zero, as all samples fell into the same haplotype. For the northern Scandinavian brown bears, haplotype and nucleotide diversity were similar before and after the bottleneck. Tajima’s D was not significant in any of the datasets. Due to the lack of segregating sites, Tajima’s D was not estimated for the post-bottleneck southern Scandinavian samples.
Discussion
In this study, we generated 41 ancient, historical, and modern Scandinavian brown bear mitogenomes and were able to further investigate the Scandinavian brown bears’ recolonisation history and the repercussions of the recent population decline. Our results suggest that Scandinavia was recolonised by several mitochondrial lineages from the south, and a single mitochondrial lineage from the north. Furthermore, we find that the recent human-induced population decline likely resulted in more severe genomic consequences for brown bears in southern compared to northern Scandinavia.
With the retreat of the Fennoscandian ice sheet, it is thought that brown bears recolonised Scandinavia from two directions. This hypothesis was based on analyses of short mitochondrial DNA sequences, where brown bears in southern Scandinavia were shown to carry mitochondrial clade 1a and in northern Scandinavia carry mitochondrial clade 3a. It is also thought that these expanding populations met in central Sweden24,32, where a contact zone was formed due to the philopatric behaviour of female brown bears43,44. Using mitogenomes, we confirm this mitochondrial phylogeographic pattern (Fig. 2), with the southern Scandinavian brown bears falling into clade 1a and the northern Scandinavian brown bears falling into clade 3a. We did not find any southern brown bears carrying clade 3a among the ancient samples, as reported by Ersmark et al.30, or clade 1b which has been reported in Denmark32. This absence can likely be due to the limited nature of the dataset. Although speculative, it is possible that clade 3a and clade 1b did not persist in southern Scandinavia after the initial recolonisation, as we do not find any evidence of them in the considerably larger historical and modern datasets either. Moreover, it may indicate that the recolonisation event of Scandinavia did not temporally overlap with the presence of clade 1b in Denmark.
During the Pleistocene/Holocene transition, there were two short periods when the south of Scandinavia was temporarily connected to Denmark and Germany via land bridges. The first one occurred between 13.1 and 12.7 kya, and the more recent one between 12.1 and 10.3 kya (after Björck45, Radiocarbon dates calibrated by Herman et al.20). Since the inundation of the second land bridge, southern Scandinavia has been disconnected from the central European mainland. Although it is unclear whether brown bears crossed during the first or the second land bridge, it should be noted that indirectly dated fossils suggest that brown bears were present in Denmark around 13.4 kya46. This indicates that the ecological flexibility of brown bears47,48 allowed them to expand northwards close to the retreating ice edge. Taken together, it is therefore possible, and perhaps likely, that brown bears crossed using the first land bridge and thus was one of the earliest species to recolonise Scandinavia, a speculation shared in previous studies27,32. Thus, ~ 13 kya should be considered as the earliest possible recolonisation date.
The number of Scandinavian brown bear mitochondrial lineages with divergence dates preceding 13 kya can thus give insight into how many female lineages were involved in the recolonisation of southern Scandinavia. Our results support a multiple female lineage scenario (Fig. 3). The first divergence among mitochondrial lineages in our Scandinavian dataset was estimated to 23.7 kya (95% HPD: 30.7–17.5 kya), which precedes the earliest possible recolonisation event. The second split was estimated to 18.3 kya (95% HPD: 24.9–12.4 kya), for which the median node age again predates 13.1 kya, but the lower range of the 95% HPD overlaps with the presence of the first land bridge. Hence, our data indicates that the south of Scandinavia was recolonised by at least two, and possibly three mitochondrial lineages. The genetic timelines suggest that the recolonisation of southern Scandinavia was not a straightforward process but possibly involved more complex migration patterns. Two plausible scenarios emerge from our data: (1) Simultaneous recolonisation: Multiple female lineages may have crossed the land bridges simultaneously during one of the available windows, suggesting a migration possibly driven by favourable environmental conditions and resource availability; (2) Sequential recolonisation: Alternatively, the region could have been colonised in phases, with different groups migrating during the separate availability of the two land bridges, indicating a staggered recolonisation process. Interestingly, it seems that during the early Holocene, more mitochondrial lineages were present in southern Scandinavia compared to today. The mitochondrial lineage leading to Sw4 (age: 10,290+-57 calibrated years before present) seems to not have left any descendants in the 19th and 20th century and present-day populations, which likely is a result of recurrent Holocene genetic drift.
In contrast, our analyses suggest that the north of Scandinavia was initially recolonised by a single mitochondrial lineage (Fig. 4), since the most recent common ancestor of the northern Scandinavian brown bears was estimated to 11.4 kya (95% HPD: 15.0–8.1 kya). This timing coincides with the retreat of the Fennoscandian ice sheet12. The presence of modern brown bears from Finland within the same clade as Scandinavian brown bears could reflect the northern connectivity. Indeed, recent gene flow between Finnish and Scandinavian male brown bears has been identified49. Further research using a temporal approach including ancient, historical, and modern samples from Sweden, Norway and Finland could provide additional insights on this matter.
It is worth acknowledging that this study relies solely on mitochondrial genomes, a single locus genetic marker with a strictly maternal mode of inheritance and no recombination. Therefore, mtDNA can only provide insight into the female population history. It is not surprising, then, that mitochondrial and nuclear genomes may yield discordant results. A recent analysis of modern nuclear genomes suggests that bears from southern and northern Scandinavia are more similar than previously thought50. Several factors, including sex-biased gene flow or a single recolonisation followed by diversification, could explain this autosomal pattern. However, given that our modern, historical and ancient mitogenomes are highly consistent with two different re-colonisation routes, we hypothesise that the observed autosomal similarity between bears from southern and northern Scandinavia in de Jong et al.50 is likely due to male-mediated gene flow during the Holocene.
Using short mitochondrial sequences, Xenikoudakis et al.39 reported a higher pre-bottleneck mitochondrial diversity in southern compared to northern Scandinavia. Our results show that this pattern likely is due to a higher number of founding lineages in southern Scandinavia.
To further investigate the impact of human persecution on the Scandinavian brown bears, we assessed the change in mitochondrial diversity across the past 100 years. We identified a high mitochondrial diversity in Scandinavian brown bears prior to the bottleneck, especially in the southern population where each pre-bottleneck individual had a unique haplotype (Fig. 5; Table 1). This differs from the results of Bray et al.32, which reported a low mitochondrial diversity in Scandinavian brown bears already since the early Holocene using short mitochondrial markers.
As a consequence of the bottleneck, all mitochondrial diversity was lost for southern Scandinavian brown bears, with only a single haplotype remaining in the contemporary population. In contrast, the northern Scandinavian brown bears retained most haplotypes (Fig. 5; Table 1). These findings are in line with the results presented in Xenikoudakis et al.39, who, based on short mitochondrial markers and microsatellites identified a reduction of mitochondrial haplotypes and allelic richness in Scandinavian brown bears following the bottleneck, which was more pronounced in the south. Moreover, lower genetic diversity in southern Scandinavia today is also in agreement with the findings in de Jong et al.50, who reported longer runs of homozygosity in southern Scandinavian brown bears compared to the north50. Furthermore, our findings of a severe loss of mitochondrial diversity in southern Scandinavian brown bears matches the expectations for bottlenecked populations. It is in agreement with reports for other species experiencing population declines, such as the Iberian lynx (Lynx pardinus)51, Western European and Iberian wolves (Canis lupus)52,53, and Central European moose (Alces alces)54. Interestingly, however, other large carnivores in Scandinavia, such as the wolverine and wolf, have been shown to have had low mitochondrial diversity already prior to their 20th century bottlenecks55,56. This suggests that the Scandinavian brown bear had a larger long-term (pre-bottleneck) effective population size compared to sympatric wolverines and wolves.
Our results suggest that the bottleneck had less impact on the northern Scandinavian brown bears. Moreover, haplotype diversity in the northern post-bottleneck Scandinavian bears is similar to that reported for post-bottleneck bears from Finland, which was speculated to be a result of population recovery due to migratory bears from Russia57. For Scandinavian brown bears, however, we suggest a different scenario. The difference in impact of the bottleneck likely mirrors the brown bear’s extermination in large parts of southern Scandinavia, and the continuous presence and larger numbers reported for brown bears in central and northern regions34,35,38. The historical pressure has led to today’s distribution of the southern lineages now restricted closer to the contact zone in Jämtland. The only haplotype found among southern modern samples of this study is a continuation of the haplotype found in a historical sample (GX0032) from this area. Thus, the southern Scandinavian brown bears likely suffered from a more severe population decline, while mitochondrial diversity in the north was maintained. A similar pattern has been reported for Scandinavian otters (Lutra lutra), in which a pronounced loss of genetic diversity was observed in the southern but not in the northern otters58.
Conclusions
In conclusion, our results contribute significantly to the existing body of knowledge on the genetic diversity of Scandinavian brown bears by focusing on mitochondrial genomes. We have provided insights into the maternal lineage and historical patterns of genetic variation. Our study adds depth to the understanding of how the postglacial recolonisation and recent population pressures have shaped the genetic diversity of brown bears in Scandinavia. However, it is worth noting that mitochondrial genomes only represent one genetic marker and that it only reflects maternal ancestry. To obtain a more nuanced picture of the postglacial history and Holocene changes in gene flow and diversity of the Scandinavian brown bear, a temporal approach using autosomal genome-wide markers is necessary.
Methods
Sampling
The dataset consists of 41 ancient, historical and modern samples from Sweden and Norway (Fig. 1; Supplementary Table S1). The three ancient bone and tooth samples were dated to ca. 7600 years before present, 10,290 ± 57 calibrated years before present, and 1916 ± 27 calibrated years before present30. The 17 historical bone and skin samples date to between the 1830’s to 1940’s, and the 21 modern muscle tissue samples were collected between 2007 and 2012. All samples were obtained from museum collections or provided by collaborators (Supplementary Table S1). All ancient and historical laboratory work was carried out in facilities dedicated for working with degraded DNA at the Museum of Natural History and the Centre for Palaeogenetics in Stockholm.
DNA extraction, library build and sequencing
Modern samples
The 21 modern samples were collected from muscle tissue and extracted using the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. The DNA extracts were sent to National Genomics Infrastructure (NGI), Stockholm for modern DNA library preparation, using the Illumina TruSeq PCR-free kit (350 bp), and sequenced on 0.5 lanes of a Illumina NovaSeq 6000 S4-300 flow-cell, with a paired-end 2 × 150 bp setup.
Historical samples
For the 17 historical samples, DNA extracts from 16 bone samples and one skin sample were obtained from a previous study39. Double stranded libraries were built following a modified version of the Meyer Kircher protocol59, with additional treatment with 6 units of USER Enzyme mix (New England Biolabs) and AccuPrime™ Pfx DNA Polymerase (Thermo Fisher Scientific Inc.). A unique indexing primer combination was used per library to ensure unique labelling. Five to six amplifications per sample were pooled and cleaned with Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA). Each cleaned pool was run on a high-sensitivity DNA chip on a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) to assess DNA concentration and fragment size distribution. All historical samples were pooled in equimolar ratios and sequenced on one lane of an Illumina HiSeq 2500 platform with a paired end 2 × 125 bp setup.
Ancient samples
DNA extracts were obtained from a previous study30. Following60, double stranded Illumina libraries were built with a modified version of the Meyer Kircher protocol59, using 3 Units of USER enzyme. DNA library products were purified with Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA). DNA concentration and fragment size distribution was measured on a high-sensitivity DNA chip on a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). One index PCR for Sw1, Sw4 and Sw9 were initially sequenced on a Illumina Novaseq 6000 S4-200 v1.5 platform, with a paired-end 2 × 100 bp setup. Sw1 and Sw4 were sequenced in a second sequencing run with the same settings, to further increase coverage. For Sw9 sufficient yield was reached from the initial sequencing run.
Data processing
For historical and modern samples, a beta-version of the GenErode pipeline was used, while ancient samples were processed using GenErode v0.4.261. GenErode is a bioinformatics pipeline to process and analyse ancient, historical, and modern genomic data61. Adapter trimming and read merging was done for modern samples using TrimGalore v0.6.5 (https://github.com/FelixKrueger/TrimGalore), for historical samples using a modified version of SeqPrep v1.1 (https://github.com/jstjohn/SeqPrep) as in62, and for ancient samples using fastp v0.22.263. Reads shorter than 30 bp were discarded. In order to avoid nuclear mitochondrial DNA segments (NUMTs), the trimmed and merged reads were mapped to the nuclear-mitochondrial polar bear (Ursus maritimus) reference genome [NCBI RefSeq accession no. GCF_017311325.1;64]. For mapping sequenced DNA reads, the BWA v0.7.17 mem algorithm65 was used for modern samples, and the aln algorithm66 with specific ancient DNA settings (deactivated seeding: -l 16500, more substitutions allowed: -n 0.01, up to two gaps allowed: -o 2) was used for historical and ancient samples62. Duplicates were marked with Picard v2.22.4 MarkDuplicates (https://broadinstitute.github.io/picard/) for modern and removed with SAMtools v1.967 for historical and ancient samples, and IndelRealigner from GATK v3.768 was used to realign reads around indels. A second round of duplicate removal using the samremovedup.py script from GenErode v0.4.2 was performed for Sw1 and Sw4, to remove remaining duplicates after merging of bam files of the same samples but different index PCRs. For Sw9 and the historical and modern samples, only one index per sample was used, thus no second round of duplicate removal was required. Mitochondrial genomes of all samples were extracted using samtools v1.9 with filtering for mapping quality (-q30), and consensus sequences were called with ANGSD v0.93369 using the majority rule and filtering for quality score (-minQ 30), mapping quality (-minmapQ 30), as well as minimum coverage of 3X per site, with ambiguous sites being called as N.
Computational analyses
Alignment
We aligned the 41 Scandinavian brown bear mitochondrial genomes in MEGA1170. Following Fortes et al.41, we excluded a section of the D loop. For phylogenetic and divergence time analyses, we furthermore excluded the Leu1, ND1, Ile, Gln and Met partitions since they consisted of unknown sites (N) only. We then aligned our 41 mitogenomes with additional 408 published mitogenomes, resulting in a 15,118 bp alignment of 449 brown and polar bears (Supplementary Table S4).
Phylogenetic relationship and divergence
A maximum clade credibility tree of 449 brown and polar bear mitogenomes was generated in BEAST v1.10.471, using a strict molecular clock, a previously estimated substitution rate of 2.48 × 10−8 mutations per site per year8, and a constant size coalescent tree model. HKY with a gamma distribution and invariant sites was selected as the nucleotide substitution model based on the Bayesian Information Criterion in Jmodeltest v2.1.1072,73. Tip dates were specified as radiocarbon dates, previously published molecular date estimates, or estimated ages based on archaeological context (Supplementary Tables S1, S4 ). The age of Sw1 was estimated by sample tip dating, using a uniform prior with a mean of 7600 years ago and a range of 0 to 50,000 years ago. We ran 100 million MCMC iterations, sampling every 5000 states. The BEAST output was verified for convergence and effective sample size of the chains in Tracer v1.7.274, and a consensus tree (with 10% burn-in removed) was generated in TreeAnnotator v1.10.4 obtained from the BEAST toolkit. The maximum clade credibility phylogenetic tree was visualised in Figtree v1.4.4 (https://github.com/rambaut/figtree/releases/tag/v1.4.4) and Linearity Curve v.5.0.0 (https://www.linearity.io/curve/). See Supplementary Figure S1 for all individual samples.
Mitogenome networks
To avoid biases, we excluded ambiguous sites (N) which resulted in a 11,412 bp alignment of the 41 Scandinavian brown bears. We generated a temporal haplotype network of the 41 full mitochondrial genomes using the R script TempNet v1.875. To enhance clarity, the haplotype network was subsequently redrawn using Linearity Curve v.5.0.0. Additionally, a median-joining haplotype network was generated in PopART with default parameters and epsilon = 076 (Supplementary Figure S2).
Mitogenome diversity
For mitochondrial genetic diversity analysis, we used the 11,412 bp alignment of the 41 Scandinavian brown bears, excluding unknown sites (N). We calculated mitochondrial genetic diversity indices including haplotype diversity (Hd), nucleotide diversity (π) and Tajima’s D77 using DNAsp v.6.12.0378. Tajima’s D was calculated excluding the three ancient samples. For this analysis, the dataset was split by haplogroup and time periods; pre-bottleneck clade 1a (n = 9), post-bottleneck clade 1a (n = 10), pre-bottleneck clade 3a (n = 11), post-bottleneck clade 3a (n = 11) (Supplementary Table S1).
Data availability
Mitochondrial sequences obtained in this study are deposited in GenBank (accession numbers PQ334008-PQ334048). The xml-file used as input for the BEAST analysis is available at the Zenodo repository (DOI: 10.5281/zenodo.13744721).
References
Hewitt, G. M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. Lond. 68, 87–112 (1999).
Hewitt, G. M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. Lond. 58, 247–276 (1996).
Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 405, 907–913 (2000).
Taberlet, P., Fumagalli, L., Wust-Saucy, A. G. & Cosson, J. F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 7, 453–464 (1998).
Stewart, J. R., Lister, A. M., Barnes, I. & Dalén, L. Refugia revisited: Individualistic responses of species in space and time. Proc. Biol. Sci. 277, 661–671 (2010).
Stewart, J. R. & Lister, A. M. Cryptic northern refugia and the origins of the modern biota. Trends Ecol. Evol. 16, 608–613 (2001).
Provan, J. & Bennett, K. D. Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 23, 564–571 (2008).
Anijalg, P. et al. Large-scale migrations of brown bears in Eurasia and to North America during the Late Pleistocene. J. Biogeogr. 45, 394–405 (2018).
Parducci, L. et al. Glacial survival of boreal trees in northern Scandinavia. Science 335, 1083–1086 (2012).
Clark, P. U. et al. The last glacial maximum. Science 325, 710–714 (2009).
Hughes, A. L. C., Gyllencreutz, R., Lohne, Ø. S., Mangerud, J. & Svendsen, J. I. The last Eurasian ice sheets—A chronological database and time-slice reconstruction, DATED-1. Boreas 45, 1–45 (2016).
Stroeven, A. P. et al. Deglaciation of Fennoscandia. Quat. Sci. Rev. 147, 91–121 (2016).
Dalén, L. et al. Ancient DNA reveals lack of postglacial habitat tracking in the arctic fox. Proc. Natl. Acad. Sci. U. S. A. 104, 6726–6729 (2007).
McDevitt, A. D., Coscia, I., Browett, S. S., Ruiz-González, A., Statham, M. J, Ruczyńska, I., et al. Next-generation phylogeography resolves post-glacial colonization patterns in a widespread carnivore, the red fox (Vulpes vulpes), in Europe. bioRxiv. 2021. p. 2020.02.21.954966. https://doi.org/10.1101/2020.02.21.954966
Fedorov, V., Jaarola, M. & Fredga, K. Low mitochondrial DNA variation and recent colonization of Scandinavia by the wood lemming Myopus schisticolor. Mol. Ecol. 5, 577–581 (1996).
Seddon, J. M., Santucci, F., Reeve, N. J. & Hewitt, G. M. DNA footprints of European hedgehogs, Erinaceus europaeus and E. concolor: Pleistocene refugia, postglacial expansion and colonization routes. Mol. Ecol. 10, 2187–2198 (2001).
Thörn, F. et al. The effects of drift and selection on latitudinal genetic variation in Scandinavian common toads (Bufo bufo) following postglacial recolonisation. Heredity 126, 656–667 (2021).
Lundqvist, A.-C., Alström-Rapaport, C. & Tegelström, H. Fennoscandian phylogeography of the common shrew Sorex araneus. Postglacial recolonisation—Combining information from chromosomal variation with mitochondrial DNA data. Acta Theriol. 56, 103–116 (2011).
Günther, T. et al. Population genomics of Mesolithic Scandinavia: Investigating early postglacial migration routes and high-latitude adaptation. PLoS Biol. 16, e2003703 (2018).
Herman, J. S. et al. Land-bridge calibration of molecular clocks and the post-glacial colonization of Scandinavia by the Eurasian field vole Microtus agrestis. PLoS One 9, e103949 (2014).
Nota, K. et al. Norway spruce postglacial recolonization of Fennoscandia. Nat. Commun. 13, 1333 (2022).
Lagerholm, V. K. et al. On the origin of the Norwegian lemming. Mol. Ecol. 23, 2060–2071 (2014).
Randi, E., Gentile, L., Boscagli, G., Huber, D. & Roth, H. U. Mitochondrial DNA sequence divergence among some west European brown bear (Ursus arctos L.) populations. Lessons for conservation. Heredity 73(Pt 5), 480–489 (1994).
Taberlet, P. & Bouvet, J. Mitochondrial DNA polymorphism, phylogeography, and conservation genetics of the brown bear Ursus arctos in Europe. Proc. Biol. Sci. 255, 195–200 (1994).
Kohn, M., Knauer, F., Stoffella, A., Schröder, W. & Pääbo, S. Conservation genetics of the European brown bear–a study using excremental PCR of nuclear and mitochondrial sequences. Mol. Ecol. 4, 95–103 (1995).
Swenson, J. E., Taberlet, P. & Bellemain, E. Genetics and conservation of European brown bears Ursus arctos. Mamm. Rev. 41, 87–98 (2011).
Sommer, R. S. & Benecke, N. The recolonization of Europe by brown bears Ursus arctos Linnaeus, 1758 after the Last Glacial Maximum. Mamm. Rev. 35, 156–164 (2005).
Saarma, U. & Kojola, I. Matrilineal genetic structure of the brown bear population in Finland. Ursus 18, 30–37 (2007).
Korsten, M. et al. Sudden expansion of a single brown bear maternal lineage across northern continental Eurasia after the last ice age: A general demographic model for mammals?. Mol. Ecol. 18, 1963–1979 (2009).
Ersmark, E. et al. Genetic turnovers and northern survival during the last glacial maximum in European brown bears. Ecol. Evol. 9, 5891–5905 (2019).
Davison, J. et al. Late-Quaternary biogeographic scenarios for the brown bear (Ursus arctos), a wild mammal model species. Quat. Sci. Rev. 30, 418–430 (2011).
Bray, S. C. E. et al. Ancient DNA identifies post-glacial recolonisation, not recent bottlenecks, as the primary driver of contemporary mtDNA phylogeography and diversity in Scandinavian brown bears. Divers. Distrib. 19, 245–256 (2013).
Taberlet, P., Swenson, J. E., Sandegren, F. & Bjärvall, A. Localization of a contact zone between two highly divergent mitochondrial DNA lineages of the brown bear Ursus arctos in Scandinavia. Conserv. Biol. 9, 1255–1261 (1995).
Swenson, J. E. et al. Size, trend, distribution and conservation of the brown bear Ursus arctos population in Sweden. Biol. Conserv. 70, 9–17 (1994).
Swenson, J. E. et al. The near extinction and recovery of brown bears in Scandinavia in relation to the bear management policies of Norway and Sweden. Wildl. Biol. 1, 11–25 (1995).
Bjärvall, A. The brown bear in Sweden: Distribution, abundance, and management. Bears Biol. Manag. 4, 255–257 (1980).
Swenson, J. E. et al. Challenges of managing a European brown bear population; Lessons from Sweden, 1943–2013. Wildl. Biol. 2017, 1–13 (2017).
Kindberg, J. & Swenson, J. E. Björnstammens storlek i Sverige 2017. Skandinaviska björnprojektet report. 2018;3. Available: https://www.slu.se/globalassets/ew/org/centrb/vsc/vsc-dokument/inventering/bjornstammens-storlek-i-sverige-2017.pdf.
Xenikoudakis, G. et al. Consequences of a demographic bottleneck on genetic structure and variation in the Scandinavian brown bear. Mol. Ecol. 24, 3441–3454 (2015).
South, A. rworldmap: A new R package for mapping global data. R J. 2011;3. Available: http://www.econ.upf.edu/~michael/visualdata/RJournal_2011-1_South.pdf.
Fortes, G. G. et al. Ancient DNA reveals differences in behaviour and sociality between brown bears and extinct cave bears. Mol. Ecol. 25, 4907–4918 (2016).
Segawa, T. et al. Ancient DNA reveals multiple origins and migration waves of extinct Japanese brown bear lineages. R. Soc. Open Sci. 8, 210518 (2021).
Støen, O.-G., Zedrosser, A., Saebø, S. & Swenson, J. E. Inversely density-dependent natal dispersal in brown bears Ursus arctos. Oecologia 148, 356–364 (2006).
Zedrosser, A., Støen, O.-G., Sæbø, S. & Swenson, J. E. Should I stay or should I go? Natal dispersal in the brown bear. Anim. Behav. 74, 369–376 (2007).
Björck, S. A review of the history of the Baltic Sea, 13.0–8.0 ka BP. Quat. Int. 27, 19–40 (1995).
Aaris-Sørensen, K. Diversity and dynamics of the mammalian fauna in Denmark throughout the last glacial-interglacial cycle, 115–0 Kyr BP. Fossils Strata, Vol. 57. 1–59 (2009).
Swenson, J. E., Gerstl, N., Dahle, B. & Zedrosser, A. Action plan for the conservation of the brown bear (Ursus arctos) in Europe. Nat. Environ. 114, 1–69 (2000).
Bojarska, K. & Selva, N. Spatial patterns in brown bear Ursus arctos diet: The role of geographical and environmental factors. Mamm. Rev. 42, 120–143 (2012).
Kopatz, A. et al. Restoration of transborder connectivity for Fennoscandian brown bears (Ursus arctos). Biol. Conserv. 253, 108936 (2021).
de Jong, M. J. et al. Range-wide whole-genome resequencing of the brown bear reveals drivers of intraspecies divergence. Commun. Biol. 6, 153 (2023).
Casas-Marce, M. et al. spatiotemporal dynamics of genetic variation in the Iberian lynx along its path to extinction reconstructed with ancient DNA. Mol. Biol. Evol. 34, 2893–2907 (2017).
Dufresnes, C., Miquel, C., Remollino, N., Biollaz, F., Salamin, N., Taberlet, P., et al. Howling from the past: Historical phylogeography and diversity losses in European grey wolves. Proc. Biol. Sci. 285. https://doi.org/10.1098/rspb.2018.1148 (2018).
Salado, I., Preick, M., Lupiáñez-Corpas, N., Fernández-Gil, A., Vilà, C., Hofreiter, M., et al. Loss of mitochondrial genetic diversity despite population growth: The legacy of past wolf population declines. Genes. 14. https://doi.org/10.3390/genes14010075 (2022).
Niedziałkowska, M. et al. Spatial structure in European moose (Alces alces): Genetic data reveal a complex population history. J. Biogeogr. 41, 2173–2184 (2014).
Walker, C. W., Vilà, C., Landa, A., Lindén, M. & Ellegren, H. Genetic variation and population structure in Scandinavian wolverine (Gulo gulo) populations. Mol. Ecol. 10, 53–63 (2001).
Flagstad, Ø. et al. Two centuries of the Scandinavian wolf population: Patterns of genetic variability and migration during an era of dramatic decline. Mol. Ecol. 12, 869–880 (2003).
Keis, M. et al. Complete mitochondrial genomes and a novel spatial genetic method reveal cryptic phylogeographical structure and migration patterns among brown bears in north-western Eurasia. J. Biogeogr. 40, 915–927 (2013).
Tison, J.-L. et al. Population structure and recent temporal changes in genetic variation in Eurasian otters from Sweden. Conserv. Genet. 16, 371–384 (2015).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, dp.prot5448 (2010).
Dehasque, M., Pečnerová, P., Kempe Lagerholm, V., Ersmark, E., Danilov, G. K., Mortensen, P. et al. Development and optimization of a silica column-based extraction protocol for ancient DNA. Genes 13. https://doi.org/10.3390/genes13040687 (2022).
Kutschera, V. E. et al. GenErode: A bioinformatics pipeline to investigate genome erosion in endangered and extinct species. BMC Bioinf. 23, 228 (2022).
Palkopoulou, E. et al. Complete genomes reveal signatures of demographic and genetic declines in the woolly mammoth. Curr. Biol. 25, 1395–1400 (2015).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Laidre, K. L. et al. Glacial ice supports a distinct and undocumented polar bear subpopulation persisting in late 21st-century sea-ice conditions. Science 376, 1333–1338 (2022).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [q-bio.GN]. 2013. Available: http://arxiv.org/abs/1303.3997.
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinf. 15, 356 (2014).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027 (2021).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat Methods. 9, 772 (2012).
Guindon, S. & Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704 (2003).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst Biol. 67, 901–904 (2018).
Prost, S. & Anderson, C. N. K. TempNet: A method to display statistical parsimony networks for heterochronous DNA sequence data. Methods Ecol. Evol. 2, 663–667 (2011).
Leigh, J. W. & Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302 (2017).
Acknowledgements
This study was funded by the Swedish Research Council grant No. 2020-04808. I.N.M has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 885088. We would like to thank our collaborators who provided samples for this study. For modern and historical samples, we would like to thank Daniela Kalthoff, Peter Mortensen and Peter Nilsson from the Swedish Museum of Natural History. We would also like to thank Friedrike Johansson and Göran Nilsson at the Natural History Museum in Gothenburg for providing historical and ancient samples. Furthermore, we would like to thank Øysten Wiig at the Natural History Museum in Oslo for providing historical samples, and Linda Wickström at Geological Survey of Sweden (SGU) in Uppsala, and Gunilla Eriksson and Markus Fjellström at the Department of Archaeology, Stockholm University for providing ancient samples. We also acknowledge computational resources (project IDs: SNIC 2022/5-27, NAISS 2023/5-22, SNIC 2022/22-468, NAISS 2023/22-577, SNIC 2022/22-280, NAISS 2023/22-762) provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at the Uppsala Multidisciplinary Centre for advanced computational Science partially funded by the Swedish Research Council through grant agreements no. 2022-06725 and no. 2018-05973. We want to thank the two reviewers and the editor for their time and insightful suggestions which helped us to improve the manuscript.
Funding
Open access funding provided by Stockholm University.
Author information
Authors and Affiliations
Contributions
L.D. and I.N.M., conceived and designed the study. I.N.M., J.v.S., G.X., E.E., and I.S.F. performed laboratory work. I.S.F. and I.N.M. undertook data analyses with input from E.L. I.S.F. and I.N.M. wrote the manuscript with input from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feinauer, I.S., Lord, E., von Seth, J. et al. Heterochronous mitogenomes shed light on the Holocene history of the Scandinavian brown bear. Sci Rep 14, 24917 (2024). https://doi.org/10.1038/s41598-024-75028-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-75028-6
