
Abstract
Season of birth, viral infections, HLA haplogenotypes and non-HLA variants are implicated in the development of celiac disease and celiac disease autoimmunity, suggesting a combined role of genes and environmental exposures. The aim of the study was to further decipher the biological pathways conveying the season of birth effect in celiac disease autoimmunity to gain novel insights into the early pathogenesis of celiac disease. Interactions between season of birth, genetics, and early-life environmental factors on the risk of celiac autoimmunity were investigated in the multicenter TEDDY birth cohort study. Altogether 6523 genetically predisposed children were enrolled to long-term follow-up with prospective sampling and data collection at six research centers in the USA, Germany, Sweden and Finland. Celiac disease autoimmunity was defined as positive tissue transglutaminase antibodies in two consecutive serum samples. There was a significant season of birth effect on the risk of celiac autoimmunity. The effect was dependent on polymorphisms in CD247 gene encoding for CD3ζ chain of TCR-CD3 complex. In particular, children with major alleles for SNP rs864537A > G, in CD247 (AA genotype) had an excess risk of celiac autoimmunity when born March–August as compared to other months. The interaction of CD247 with season of birth on autoimmunity risk was accompanied by interactions with febrile infections between the ages of 3–6 months. Considering the important role of TCR-CD3 complex in the adaptive immune response and our findings here, CD247 variants and their possible effect of subgroups in autoimmunity development could be of interest in the design of future gene-environment studies of celiac disease. ClinicalTrials.gov Identifier: NCT00279318.
Similar content being viewed by others

Introduction
Celiac disease (CD) is a systemic immune-mediated disorder characterized by the presence of serum autoantibodies. Prospective birth cohort studies with longitudinal screening have revealed the highest incidence between the age of one and four years, although the disease can manifest at any age after gluten introduction1. Development of CD and CD autoimmunity (CDA) requires the presence of HLA-DQ2/DQ8 genotypes2, with additional influence of at least 40 non-HLA loci3,4,5. Although the genetic risk is present in ~ 40% of White people, only a minority of the predisposed individuals are affected, and even genetically identical monozygotic twins show only partial concordance6,7. A rapid change in the incidence in many countries8, together with the varying prevalence between genetically similar populations in nearby geographical regions9, suggest a role of environmental factors in the etiology. Previous evidence indicates that e.g., the amount of gluten intake and exposure to viral infections in early childhood could constitute such factors10,11,12,13,14,15.
Further indication for the role of environment comes from the unexplained fluctuations in CD incidence depending on the season of birth (SoB), most studies finding spring and summer birth being associated with higher risk compared to fall and winter14,16,17,18,19,20. Seasonal variation has also been reported in respect to other autoimmune-mediated diseases such as Crohn’s disease and ulcerative colitis21, juvenile idiopathic arthritis22, Addison’s disease23 and type 1 diabetes (T1D)24. The SoB effect is interesting, as it likely reflects an underlying biological process. For example, Sailani et al. identified distinct patterns of seasonal fluctuations across molecules and biological pathways involved in inflammation and immunity, such as the complement system and acute phase response, as well as observed that the gut microbiome undergoes seasonal changes25. Seasonality in human infectious diseases is well-established and could impact the observed fluctuations. Accordingly, our previous observation that increased CD risk due to gastrointestinal (GI) infections is influenced by SoB and modified by HLA, gluten and breastfeeding14 imply complex gene-environment interactions. SoB-related variation in immune functions such as fluctuations in innate and adaptive cytokine responses and immune cell counts is present already in cord blood and neonatal mucosal immune system, indicating the possibility of additional prenatal effects such as maternal exposures during pregnancy26,27,28. No study has yet investigated interactions between SoB and pregnancy and/or delivery-related factors on the risk of CD/CDA.
Prospective studies focused on early life provide an opportunity to elucidate the gene-environment interactions evoking autoimmunity29. The Environmental Determinants of Diabetes in the Young (TEDDY) is a multicenter birth cohort study following children at risk for T1D and CD. Prospective data collection and sampling enabled us to study interactions between SoB, genetics and environmental factors in the risk of CDA.
Materials and methods
Participants and study design
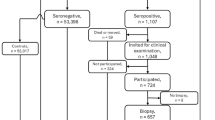
TEDDY is an observational study following children at-risk for T1D and CD from birth to the age of 15 years6. The aim was to find environmental factors triggering autoimmunity. It includes three study centres in both the USA and Europe.
Inclusion criteria have been described elsewhere31. The reasons for exclusion were unsuccessful re-contacting or failure to schedule the first appointment, lacking HLA data, disease or birth defect that disallowed follow-up or involved a treatment that might interact with the outcomes, child being never tested for CDA, first testing occurring after the age of 36 months and ineligible HLA.
Between 2004–2010, screening for HLA risk were performed on 424,788 infants of whom 21,583 carried the haplogenotypes of interest. Altogether 8,676 families joined the study, and 6,523 children were screened for CDA and followed up until 10 years of age. Of them, 1,262 (19.4%) developed CDA. The median age for seropositivity was 3.3 years and the earliest seroconversion occurred at the age of 10 months.
The study visits occurred quarterly until the age of 4 years and biannually thereafter. Early life data, including maternal and infant illnesses, was obtained from questionnaires given 3 months postpartum. Data about later infections and gluten introduction was obtained from diaries updated by the caregivers. For illnesses, caregivers recorded the nature and date of appearance and the diagnosis.
Serological measurements and outcomes
Blood samples were drawn during every visit. Annual screening for transglutaminase antibodies (tTGA) started from 24 months of age6,32. If a child was seropositive, previous samples were tested retrospectively. CDA was the primary endpoint and was defined as positive tTGA in consecutive samples ≥ 3 months apart. Seropositive children were referred for endoscopy outside the study protocol6,33.
Infections
Maternal infections during pregnancy and child’s infections were categorized as upper/lower respiratory tract infections, GI infections and other34. The child’s infections after the first 3 months were collected at each visit and further translated into ICD-10 codes. The infectious episodes were examined up to age of 12 months.
Statistical analyses
The seasons were defined as spring (March–May), summer (June–August), fall (September–November) and winter (December–February). The association of SoB with risk of CDA were tested using Proportional Hazard models. Children who were negative for CDA were censored on the time of the last tTGA test result or before the age of 10 years. All models were adjusting for HLA haplogenotypes, sex and country. The strength of relations is given by hazard ratios (HR) and 95% confidence intervals (CI). Interactions were evaluated on the ratio scale (multiplicative interaction), or by including a cross-product term in the hazard model, or on the difference scale (additive interaction), by approximating relative excess risk due to interaction (RERI) by adding in the model a 4-category variable describing the occurrence (1) and absence (0) of specific time of birth and genetic factor and estimating the RERI as HR11 – HR10—HR01 + 1 (25). The RERI and 95% CI assess the additional risk due to interaction with RERI > 0 indicating synergistic interaction and RERI < 0 implying antagonistic interaction. The strongest interactions were considered to occur on both the additive and multiplicative scale. P-values < 0.05 were considered significant.
Genetic analyses
SNPs were genotyped using the ImmunoChip and/or the T1DExomeChip (an array with > 90,000 custom content SNPs added to the Infinium® CoreExome-24 v.1.1 BeadChip (Illumina, CA)) at the Center for Public Health Genomics at the University of Virginia. Quality control measures involved ignoring subjects with a call rate < 95% or discordance with reported sex, and SNPs with a call rate < 95%, deviating from Hardy–Weinberg equilibrium in controls with the European ancestry, or with concordance < 99% in duplicates, as described previously35. To investigate interactions outside the HLA region, 87 non-HLA SNPs listed in TEDDY5 and available from ImmunoChip/TEDDY-T1DExomeChip data35 were studied for their association with CDA. The Eagle2 software Version 2.4.1 (https://alkesgroup.broadinstitute.org/Eagle/) (Loh P-R—PMID: 27,694,958) was used to estimates haplotype phase of 4 SNPs (rs840016, rs864537, rs2056626, and rs1723016). Associated SNPs (p-value < 0.05) were selected for interaction testing with spring/summer birth on both additive and multiplicative scale at 5/10 years of age accounting for population stratification (ancestral heterogeneity) (utilizing the top 3 principal components approximated from the T1DExomeChip). Haploview (Version 4.2) was used to generate the linkage disequilibrium (LD) plot.
To study for possible functional role of genes interacting with SoB, mRNA expression from whole-blood samples using RNA-Seq were examined up to age 12 months. The data were available from two TEDDY case–control studies and comprised of 12% of the participants36. No difference in SoB was observed between children with and without expression data (Supplementary Table 1). Differences in expression across CD247 genotypes were tested at 3, 6, 9 and 12 months of age with adjustment for country, sex, HLA, first-degree relative with T1D and presence of islet autoantibodies.
The TEDDY RNA-sequencing (RNA-seq) samples were prepared using Illumina’s TruSeq Stranded mRNA Sample Prep Kit. Sequence were created on the Illumina HiSeq4000 platform using paired-end 2 × 101 bp reads with a targeted 50 million reads per sample by the Broad Institute (Cambridge, MA). The raw sequences were aligned against the Gencode Genome Reference Consortium Build 38 (GRCh38.p12) Release 31 reference genome37 and comprehensive gene annotation. Reads were aligned using the STAR aligner (version 2.6.1d)38 based on the Trans-Omics for Precision Medicine GTEx RNA-Seq analysis pipeline39. Ribosomal RNA (rRNA) was identified and removed using BBSplit version 38.86 (https://sourceforge.net/projects/bbmap/) prior to alignment. Ensembl release 9740 was used as reference.
Gene expression was calculated using RNA-SeQC (version 2.1.0)41 and normalized using the Total Ubiquitous (TU) method from the NormExpression R package (version 0.1.0)42. The ubiquitous genes were selected by the Total Ubiquitous method in the NormExpression R package. The code for this can be found in Sect. 1D of the documentation for the package (https://github.com/cran/NormExpression/blob/master/inst/doc/readme.pdf). For the TEDDY RNA-seq data, it was run using a pre ratio = 1, lower_trim = 0.4, and upper_trim = 0.95. To account for changes in sampling protocol over time, for each gene an RNA Quality Score (RQS) correction was applied to non-zero TU normalized levels by fitting a linear spline with a knot at the 7.5 RQS value. To ensure the RQS adjusted values for samples with RQS > = 7.5 remained nonnegative and were similarly centered to the original TU normalized data, the original TU normalized values were centered at the 7.5 RQS value before running the linear spline and all adjusted RQS values were shifted to align with the minimum of the original TU normalized value for the samples with RQS ≥ 7.5. Any adjusted RQS values less than zero for samples with RQS < 7.5 were set to zero and were assumed to be too degraded to be considered having any gene expression signal or level. Expression levels were provided as transcripts per million.
Statistical analyses were performed using SAS Software Version 9.4 (https://support.sas.com/software/94/) (SAS Institute, Cary, North Carolina, U.S.A.) and R (Version 4.0.2).
Data availability
Data (https://doi.org/https://doi.org/10.58020/y3jk-x087) reported here can be obtained for request at the NIDDK Central Repository website, Resources for Research (R4R), https://repository.niddk.nih.gov/. The TEDDY RNA Sequencing data that provide support for the findings of the present study have been added in NCBI’s database of Genotypes and Phenotypes (dbGaP) with access code phs001442.v4.p3.
Results
Season of birth and risk of CDA
The number of births across months of the year ranged from 504–586 (Table 1). The 0–9-year incidence of CDA ranged from 20.1–31.9 per 1000 person-years but clustered with high incidence between April and July (Table 1). Spring/summer SoB was associated with an increased likelihood of CDA by 10 years of age (HR 1.29, 95% CI 1.09–1.53, p = 0.004) and less so by 5 years of age (1.18, 1.03–1.34, p = 0.01) after adjusting for HLA, sex or country. No significant interactions by the adjusting factors were observed with SoB (data not shown).
Interactions between SoB and non-HLA variants
After accounting for country, sex and HLA, a total of 16 SNPs previously listed in TEDDY4 were associated (p < 0.05, false discovery rate < 0.20) with CDA at 5 and 10 years of age (Supplementary Table 2). Of these, rs864537 in CD247 exhibited a significant interaction with SoB, both on the additive (p = 0.003) and multiplicative scale (p = 0.001), on the risk of CDA. The association of a spring/summer birth with CDA risk was significantly different between children with and without the minor G allele (Fig. 1). A positive correlation was seen only for children having CD247-AA genotype and an excess risk was seen only for children with this genotype and born in spring/summer (RERI additive scale − 0.46, 95% CI − 0.76 to − 0.16 and multiplicative scale 0.69, 0.55–0.87).

Absolute incidence of CDA among children with and without CD247 polymorphism as shown by season of birth (panel A), birth cohort (panel B) and age of CDA development (panel C and D).
An examination of the absolute risk of CDA by MoB and year of birth as well as by age of seroconversion showed an excess risk of CDA only for children carrying the CD247-AA genotype born in spring/summer compared to children born in winter/fall or carrying the G allele (Fig. 1A). The spring/summer effect among children with CD247-AA was consistent by year of birth (Fig. 1B) and age of seroconversion (Fig. 1A, C and D) although absolute risk changed by age and birth cohort (data not shown). The risk of CDA for children with CD247-AA was consistently higher for every MoB in spring/summer compared to fall/winter (Supplementary Fig. 1A/B). There were no significant associations between MoB or SoB and CDA among children with CD247-AG (Supplementary Fig. 1C/D) and GG (Supplementary Fig. 1E/F).
Next, the SoB by CD247 within different risk groups identified in TEDDY for CDA was examined. The relative HR of CDA for children born in the spring/summer compared to fall/winter was consistently higher for those with CD247-AA, i.e. a spring/summer birth was associated with an increased risk of CDA in every subgroup except for those who started gluten after 6 months of age (Fig. 2). SoB showed no association with CDA in any of the stratified groups of children who carried the CD247-G allele.

Hazard ratios (± 95% confidence intervals) of spring/summer season of birth on risk of CDA by 10 years of age shown by CD247-G polymorphism and adjusted a for HLA haplogenotypes, country of residence, sex of child and age when child started gluten. a = included in model if not stratified.
Other SNPs in CD247
The ImmunoChip contained 35 SNPs in CD247 in addition to rs864537 (Supplementary Fig. 2A). Four of these were excluded from further analysis as the minor allele frequency was less than 2.5%. Of the remaining SNPs, 5 out of 31 (rs840016, rs2056626, rs1723016, rs2995093, rs2982479) showed evidence of interaction with SoB on risk of CDA (Supplementary Fig. 2B), of which 3 (rs840016, rs2056626 and rs1723016) were in linkage disequilibrium (LD) with rs864537. A haplotype analysis (rs840016, rs864537, rs2056626, rs1723016) showed that SoB was associated with CDA risk (HR 1.55, 95% CI 1.27–1.89, p < 0.0001) only if the haplotype comprising of major alleles from all four SNPs was present as homozygous (CATT/CATT, Supplementary Table 3). The interaction of SoB with rs2995093 on CDA risk was no longer significant when adjusting for SoB and rs864357 interaction, whereas the interaction of SoB with rs2982479, further away from rs864537, remained associated. Further analysis revealed that the minor G allele frequency of rs2982479 was low (6.3%), but the allele was associated with a higher risk of CDA (1.51, 1.12–2.04, p = 0.007) for children born in the fall and not in other seasons.
mRNA expression
Data of mRNA expression from whole blood during first year of life were available to analyze CD247 expression in 777 children. The distribution of CD247 variants and SoB was similar for children with and without available expression data (Supplementary Table 1). The expression of CD247 was found to be lower until 6 months for children with the AA genotype in rs864537 compared to those with the G allele (Fig. 3A). Furthermore, haplotype analysis of rs840016, rs864537, rs2056626 and rs1723016 showed that the association was stronger for children with homozygous CATT haplotype (major alleles only) than those with TGGC minor allele only haplotype (Fig. 3B). Differences in expression were most prominent between 3–6 months of age (Fig. 3).

CD247 mRNA gene expression Log2 TPM levels (± 95% confidence intervals) during first year of life as shown by CD247-G polymorphism of SNP-rs864537 (Panel A) and haplogenotypes CATT/CATT or haplogenotype TGGC of SNPs rs840016, rs864537, rs2056626, rs1723016 (Panel B). Differences in means at each time point tested with adjustment for number of time points (p < 0.01) and for country of residence, sex, family history with type 1 diabetes (matching factors in the source Nested Case Control study) as well as HLA and positive for islet autoantibodies (outcome in source Nested Case Control Study).
Gene-environment interactions
Febrile respiratory and GI infectious episodes between 3–6 months of age interacted both on additive and multiplicate scale with CD247 on CDA risk by age of 5 years (additive p = 0.008; multiplicative, p = 0.007; Supplementary Table 4) and less so by age 10 years (both additive and multiplicative, p = 0.03). Other infections during pregnancy interacted with CD247-AA genotype on CDA risk by age 5 years (both additive and multiplicative p = 0.02) and 10 years (both p = 0.02), maternal urinary system infections and cold sores explaining these interactions (Supplementary Table 4). Additionally, febrile GI and respiratory infectious episodes in winter/spring between the age of 3–6 months were associated with lower incidence of CDA among CD247-G (Fig. 4), but they alone did not explain the SoB correlation with CDA risk (Fig. 4).

Absolute incidence of CDA from 0 to 4 years of age by CD247 genotype and seasonal infections reported between age 3 and 6 months of age.
After adjusting for country of residence, sex, HLA and family history of CD at age 9 months and for non-HLA risk SNPs (Supplementary Table 2), a spring or summer birth remained associated with CDA risk at 5 years of age (HR 1.38, 95% CI 1.13–1.70, p = 0.002) among children with CD247 AA. After additional adjustment for seasonal respiratory or GI infections between 3–6 months of age, the SoB association with CDA risk by 5 years of age was reduced (1.29, 0.97–1.71, p = 0.08). Respiratory or GI infections reported between 3–6 months of age, specifically in August (1.59, 1.08–2.34, p = 0.02) and September (1.58, 1.13–2.21, p = 0.007), remained associated with CDA by 5 years of age among children with CD247-AA. A similar association was seen at 10 years (data not shown).
Discussion
To date, no study has explicitly explained why SoB affects the risk for CD/CDA16,17,19,20,43. Although the differences were relatively small, it was shown that the SoB effect on the risk of CDA is synergistically linked with rs864537 in CD247 as well as with three additional SNPs rs840016, rs2056626 and rs1723016, all in LD. Particularly, children homozygous for the haplotype comprising solely of major alleles of SNPs were at a higher risk of CDA when born during spring/summer (March-August) compared to other seasons/months, whereas this was not observed among those with a haplotype with minor alleles. We also found some variation in the mRNA expression levels of CD247, which could indicate differences in early immune functions depending on distinct genetic features.
CD247 encodes for T-cell surface glycoprotein CD3ζ, a component of the TCR-CD3-complex, which is essential in the adaptive immune response coupling antigen recognition to intracellular signal-transduction pathways30. Aberrant T-cell responses have been linked to CD, and defective expression of CD247 as well as CD3ζ polymorphisms or downregulation may contribute to autoimmunity in T1D, systemic lupus erythematosus (SLE) and thyroidal disease30,44,45,46,47. Furthermore, CD3ζ may play a role in the thymic T-cell selection48. Based on murine models, mutations in CD247 can also impair thymic development of T-cells, revealing the importance of CD3ζ in T-cell development and activation49. Recent findings in vitro suggest that mutations in CD247 can impair proper TCR assembly and lead to defective function of the receptor signaling that might be associated with immunodeficiency and autoimmunity50. Pathways including CD247 have also been linked to antitumor immunity51.
Previous evidence indicates that the mRNA expression of CD247 is lower in children with CD52, further directing towards its potential role in autoimmunity. The expression has also been reported to be reduced in individuals with AA genotype in rs864537 and the minor allele G to be less abundant in celiac patients compared to A allele4. In line, here children carrying the AA genotype had lower levels of mRNA expression than those with AG/GG genotypes. Regarding the SNPs in LD with rs864537, rs840016 polymorphisms have been linked to methylation changes and associated with T1D and dermatomyositis53 and rs2056626 has been associated with systemic sclerosis54,55. Here, a haplotype analysis revealed that SoB was associated with CDA risk only if the haplotype with major alleles existed as homozygous (CATT/CATT), indicating that fine mapping studies in CD247 region are needed to identify the causal variant.
Of note, neonate gene expression is distinct from that of adults and is specific to a given cell type and stimulatory condition. There appears to be an overlap of variants linked to immune-mediated conditions and those which impact on gene expression in neonatal immune cells. Based on Mendelian randomization, changes in neonatal gene expression may have causal effects on autoimmunity risk56. Details of the variations in the expression profiles of CD247 and their possible influence in the evolvement of CD remain to be deciphered, but for example in SLE, defective expression of CD247 and a subsequent T-cell-dysfunction has been suggested to promote autoimmunity due to failure in immune tolerance55.
Maternal infections and childhood febrile respiratory/GI-infections were also associated with CDA depending on the presence of the rs864537 G-allele. Moreover, the incidence of CDA in children reporting febrile infections at 3–6 months of age varied according to CD247 rs864537 genotype. Notably, genotype-dependent differences in the expression of CD247 were most prominent at 3–6 months of age in children with CD247-AA compared to other rs864537 genotypes and, similarly, in subjects with homozygous haplotype comprising of only major alleles for rs840016, rs864537, rs20256626 and rs1723016 (children with CATT/CATT).
The association between childhood and maternal infections and CD is controversial10,11,12,13,14,57,58,59,60,61,62,63,64. Recent evaluation of information gained from prospective studies on genetic and environmental risk of CD points especially to the role of enterovirus and rotavirus infections1. Besides proposed direct and indirect effects of viral or bacterial agents64, season-dependent epigenetic programming could occur during pregnancy. For instance, according to the Developmental Origins of Health and Disease hypothesis, fetal environment has an important role in development of diseases in postnatal life. The programming could happen through epigenetic changes such as DNA-methylation, histone modification and activation or silencing of noncoding RNA-associated genes. SoB has already been associated with DNA-methylation enriched among networks relating to development, the cell cycle and apoptosis65,66. Temporal patterns have also been observed in some placenta-mediated pregnancy complications such as hypertensive disorders during pregnancy67. The role of seasonal fluctuations in immune function is supported by the variable upregulation of genes involved in proinflammatory responses and fluctuating number of immunologically important blood cells across seasons68. Moreover, SoB-related variation in neonatal immunity such as season-dependent overall leukocyte count and cell-subset variation as well as alternating cytokine and chemokine levels has been observed26.
The association between SoB and CDA was present only if gluten was introduced before the age of 7 months. While age of first gluten exposure was not associated with CDA in TEDDY69, high intake during first years of life predisposed to increased risk15. Furthermore, concomitant exposures to gluten and infections together with early cessation of breastfeeding seem to have synergistic effects14,70. The latter might be due to the beneficial effects of breastfeeding on microbiota65, although it was not an independent protective factor in TEDDY71,72. Notably, significant variations in the abundance of particular taxa and microbiome diversity across seasons have been observed, possibly due to changes in the dietary nutrient and fiber composition72,73. The observation that HLA alleles influence the microbiota74 further encourages investigation of gene-environment interactions in autoimmunity.
Strengths and limitations
Our main strength is the prospective design with an exceptionally large birth cohort. The results were also consistent between countries, and the long follow-up time enabled to explore both early and delayed exposures. Inclusion of expression analysis extended the scope beyond genetic associations, although this was possible only for a small subgroup. Self-reporting of early infections and gestational factors is a limitation; however, it might even better cover episodes not requiring healthcare contact. Further limitation is that the study comprised of at-risk children of predominantly white background. An additional factor when considering the generalizability of the results is that the inclusion criteria of the TEDDY study are initially designed to evaluate the risk of T1D, CD and CDA being secondary outcomes. Consequently, the study design does not capture a part of the known HLA-DQ risk alleles for CD. However, early screening for CD autoantibodies is systematically included in the study protocol and thus strengthens the design compared to other prospective studies with T1D as the primary outcome.
Conclusions and potential directions for future research
In conclusion, there was an increased risk of CDA in children born in spring/summer (March–August). This association was present only in children with CD247-AA genotype of the rs864537 variant in CD247 encoding for CD3ζ chain of TCR-CD3-complex and in children homozygous for a haplotype comprised of only major alleles of three additional SNPs rs840016, rs2056626 and rs1723016 in LD. Children with the rs864537 AA genotype and homozygous for the CATT haplotype also expressed reduced levels of CD247 at 3–6 months of age. Information obtained from the present study can contribute to providing a foundation for risk modelling and biomarker discovery that could be exploited in designing future studies aiming to improve our understanding of CD and other autoimmune diseases.
Data availability
Data (https://doi.org/https://doi.org/10.58020/y3jk-x087) reported can be obtained for request at the NIDDK Central Repository (NIDDK-CR) website, Resources for Research (R4R), https://repository.niddk.nih.gov/. The TEDDY RNA Sequencing (RNA-Seq) data that support the present findings have been deposited in NCBI’s database of Genotypes and Phenotypes (dbGaP); accession code phs001442.v4.p3.
Abbreviations
- CDA:
-
Celiac disease autoimmunity
- CD:
-
Celiac disease
- CD3ζ:
-
Cluster of differentiation 3 zeta chain
- CI:
-
Confidence interval
- GI:
-
Gastrointestinal
- HLA:
-
Human leukocyte antigen
- HR:
-
Hazard ratio
- MoB:
-
Month of birth
- SoB:
-
Season of birth
- SPN:
-
Single nucleotide polymorphism
- TCR:
-
T-cell receptor
- TEDDY:
-
The Environmental Determinants of Diabetes in the Young
- tTGA:
-
Tissue transglutaminase antibodies
- RERI:
-
Relative excess risk due to interaction
References
Stahl, M. et al. Coeliac disease: what can we learn from prospective studies about disease risk?. Lancet Child Adolesc Health. 8(1), 63–74. https://doi.org/10.1016/S2352-4642(23)00232-8 (2024).
Sollid, L. M. et al. Evidence for a primary association of celiac disease to a particular HLA-DQ α/β heterodimer. J Exp Med 169(1), 345–350 (1989).
Trynka, G. et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet 43(12), 1193–1201 (2011).
Dubois, P. C. et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 42(4), 295–302 (2010) (Erratum in: Nat Genet.2010 May;42(5):465).
Sharma, A. et al. Identification of Non-HLA Genes Associated with Celiac Disease and Country-Specific Differences in a Large, International Pediatric Cohort. PLoS One 11(3), e0152476 (2016).
Liu, E. et al. Risk of Pediatric Celiac Disease According to HLA Haplotype and Country. N Engl J Med 371(1), 42–49 (2014) (Erratum in: N Engl J Med. 2014 Jul 24;371(4):390).
Hervonen, K. et al. Concordance of Dermatitis Herpetiformis and Celiac Disease in Monozygous Twins. J Invest Dermatol. 115(6), 990–993 (2000).
Kang, J. Y., Kang, A. H. Y., Green, A., Gwee, K. A. & Ho, K. Y. Systematic review: worldwide variation in the frequency of coeliac disease and changes over time. Aliment Pharmacol Ther 38(3), 226–245 (2013).
Kondrashova, A. et al. Lower economic status and inferior hygienic environment may protect against celiac disease. Ann Med 40(3), 223–231 (2008).
Kårhus, L. L. et al. Influenza and risk of later celiac disease: a cohort study of 2.6 million people. Scand J Gastroenterol 53(1), 15–23 (2018).
Mårild, K., Kahrs, C. R., Tapia, G., Stene, L. C. & Størdal, K. Infections and risk of celiac disease in childhood: a prospective nationwide cohort study. Am Gastroenterol 110(10), 1475–1484 (2015).
Sandberg-Bennich, S., Dahlquist, G. & Källén, B. Coeliac disease is associated with intrauterine growth and neonatal infections. Acta Paediatr 91(1), 30–33 (2002).
Myléus, A. et al. Early infections are associated with increased risk for celiac disease: An incident case-referent study. BMC Pediatr 19(12), 194 (2012).
Kemppainen, K. M. et al. Factors that increase risk of celiac disease autoimmunity after a gastrointestinal infection in early life. Clin Gastroenterol Hepatol 15(5), 694-702.e5 (2017).
Andrén Aronsson, C. et al. Association of gluten intake during the first 5 years of life with incidence of celiac disease autoimmunity and celiac disease among children at increased risk. J Am Med Assoc 322(6), 514–523 (2019).
Capriati, T., Francavilla, R., Castellaneta, S., Ferretti, F. & Diamanti, A. Impact of the birth’s season on the development of celiac disease in Italy. Eur J Pediatr 174(12), 1657–1663 (2015).
Lebwohl, B., Green, P. H. R., Murray, J. A. & Ludvigsson, J. F. Season of birth in a nationwide cohort of coeliac disease patients. Arch Dis Child 98(1), 48–51 (2013).
Lewy, H., Meirson, H. & Laron, Z. Seasonality of birth month of children with celiac disease differs from that in the general population and between sexes and is linked to family history and environmental factors. J Pediatr Gastroenterol Nutr. 48(2), 181–185 (2009).
Ivarsson, A., Hernell, O., Nyström, L. & Persson, L. Å. Children born in the summer have increased risk for coeliac disease. J Epidemiol Community Health 57(1), 36–39 (2003).
Tanpowpong, P. et al. Multicenter study on season of birth and celiac disease: Evidence for a new theoretical model of pathogenesis. J Pediatr 162(3), 501–504 (2013).
Mikulecký, M. & Cierna, I. Seasonality of births and childhood inflammatory bowel disease. Wien Klin Wochenschr. 117(15–16), 554–557 (2005).
Berkun, Y., Lewy, H., Padeh, S. & Laron, Z. Seasonality of birth of patients with juvenile idiopathic arthritis. Clin Exp Rheumatol. 33(1), 122–126 (2015).
Pazderska, A. et al. Impact of Month of Birth on the Risk of Development of Autoimmune Addison’s Disease. J Clin Endocrinol Metab. 101(11), 4214–4218. https://doi.org/10.1210/jc.2016-2392 (2016).
Kahn, H. S. et al. Association of type 1 diabetes with month of birth among U.S. youth: The SEARCH for Diabetes in Youth Study. Diabetes Care. 32(11), 2010–2015 (2009).
Sailani, M. R. et al. Deep longitudinal multiomics profiling reveals two biological seasonal patterns in California. Nat Commun 11(1), 4933 (2020).
Thysen, A. H. et al. Season of birth shapes neonatal immune function. J Allergy Clin Immunol 137(4), 1238-1246.e13 (2016).
Gold, D. R. et al. Parental characteristics, somatic fetal growth, and season of birth influence innate and adaptive cord blood cytokine responses. J Allergy Clin Immunol 124(5), 1078–1087 (2009).
Keski-Nisula, L. et al. Production of interleukin-5, -10 and interferon-γ in cord blood is strongly associated with the season of birth. Clin Exp Allergy. 40(11), 1658–1668 (2010).
Craig, M. E. et al. Early-life factors contributing to type 1 diabetes. Diabetologia 62(10), 1823–1834 (2019).
Baniyash, M. TCR ζ-chain downregulation: Curtailing an excessive inflammatory immune response. Nat Rev Immunol 4(9), 675–687 (2004).
Lernmark, B. et al. Enrollment experiences in a pediatric longitudinal observational study: The Environmental Determinants of Diabetes in the Young (TEDDY) study. Contemp Clin Trials 32(4), 517–523 (2011).
Hoffenberg, E. J. et al. Transglutaminase antibodies in children with a genetic risk for celiac disease. J Pediatr 137(3), 356–360 (2000).
Husby, S. et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr 54(1), 136–160 (2012) (Erratum in: J Pediatr Gastroenterol Nutr. 2012 Apr;54(4):572).
Lynch, K. F. et al. Gestational respiratory infections interacting with offspring HLA and CTLA-4 modifies incident β-cell autoantibodies. J Autoimmun 86, 93–103 (2018).
Törn, C. et al. Telomere length is not a main factor for the development of islet autoimmunity and type 1 diabetes in the TEDDY study. Sci Rep. 12(1), 4516 (2022).
Lee, H. S., Lynch, K. F. & Krischer, J. P. Nested case-control data analysis using weighted conditional logistic regression in The Environmental Determinants of Diabetes in the Young (TEDDY) study: A novel approach. Diabetes Metab Res Rev 36(1), e3204 (2020).
Frankish, A. et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47(D1), D766–D773 (2019).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29(1), 15–21 (2013).
GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 369(6509), 1318–1330 (2020).
Cunningham, F. et al. Ensembl 2019. Nucleic Acids Res 47(D1), D745–D751 (2019).
DeLuca, D. S. et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28(11), 1530–1532 (2012).
Wu, Z. et al. Normexpression: An R package to normalize gene expression data using evaluated methods. Front Genet 30(10), 400 (2019).
Namatovu, F., Lindkvist, M., Olsson, C., Ivarsson, A. & Sandström, O. Season and region of birth as risk factors for coeliac disease a key to the aetiology?. Arch Dis Child 101(12), 1114–1118 (2016).
Holmberg, D., Ruikka, K., Lindgren, P., Eliasson, M. & Mayans, S. Association of CD247 (CD3ζ) gene polymorphisms with T1D and AITD in the population of northern Sweden. BMC Med Genet 17(1), 70 (2016).
Lundholm, M. et al. Variation in the Cd3ζ ( Cd247) Gene Correlates with Altered T Cell Activation and Is Associated with Autoimmune Diabetes. J Immunol 184(10), 5537–5544 (2010).
Sollid, L. M. & Jabri, B. Triggers and drivers of autoimmunity: Lessons from coeliac disease. Nat Rev Immunol 13(4), 294–302 (2013).
Takeuchi, T. & Suzuki, K. CD247 variants and single-nucleotide polymorphisms observed in systemic lupus erythematosus patients. Rheumatol (Oxford) 52(9), 1551–1555 (2013).
Yamazaki, T. et al. A Shift from Negative to Positive Selection of Autoreactive T Cells by the Reduced Level of TCR Signal in TCR-Transgenic CD3ζ-Deficient Mice. J Immunol 158(4), 1634–1640 (1997).
Jin, Y. et al. Alternatively spliced variants of murine CD247 influence T cell development and activation, revealing the importance of the CD3ζ C-terminal region. J Immunol. 212(4), 541–550 (2024).
Briones, A. C. et al. Nonsense CD247 mutations show dominant-negative features in TCR expression and function. J Allergy Clin Immunol. https://doi.org/10.1016/j.jaci.2024.06.019 (2024).
Wu, Y. et al. Disrupting the phase separation of KAT8-IRF1 diminishes PD-L1 expression and promotes antitumor immunity. Nat Cancer. 4(3), 382–400 (2023).
Lahdenperä, A. I., Fälth-Magnusson, K., Högberg, L., Ludvigsson, J. & Vaarala, O. Expression pattern of T-helper 17 cell signaling pathway and mucosal inflammation in celiac disease. Scand J Gastroenterol 49(2), 145–156 (2014).
Saraeva, R. et al. CD3Z polymorphisms and promoter hypermethylation in dermatomyositis - the role of cytosine-phosphate-guanine-related single nucleotide polymorphisms. Immunol Invest 51(4), 817–825 (2022).
Radstake, T. R. D. J. et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat Genet 42(5), 426–429 (2010).
Dieudé, P. et al. Independent replication establishes the CD247 gene as a genetic systemic sclerosis susceptibility factor. Ann Rheum Dis 70(9), 1695–1696 (2011).
Huang, Q. Q. et al. Neonatal genetics of gene expression reveal potential origins of autoimmune and allergic disease risk. Nat Commun 11(1), 3761 (2020).
Canova, C. et al. Association of maternal education, early infections, and antibiotic use with celiac disease: A population-based birth cohort study in northeastern Italy. Am J Epidemiol 180(1), 76–85 (2014).
Kahrs, C. R. et al. Enterovirus as trigger of coeliac disease: nested case-control study within prospective birth cohort. BMJ Bri Med J 13(364), l231 (2019).
Bouziat, R. et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 356(6333), 44–50 (2017).
Mårild, K., Stephansson, O., Montgomery, S., Murray, J. A. & Ludvigsson, J. F. Pregnancy outcome and risk of celiac disease in offspring: A nationwide case-control study. Gastroenterol 142(1), 39-45.e3 (2012).
Mårild, K., Kahrs, C. R., Tapia, G., Stene, L. C. & Størdal, K. Maternal Infections, Antibiotics, and Paracetamol in Pregnancy and Offspring Celiac Disease: A Cohort Study. J Pediatr Gastroenterol Nutr 64(5), 730–736 (2017).
Tjernberg, A. R. & Ludvigsson, J. F. Children with celiac disease are more likely to have attended hospital for prior respiratory syncytial virus infection. Dig Dis Sci 59(7), 1502–1508 (2014).
Auricchio, R. et al. Respiratory Infections and the Risk of Celiac Disease. Pediatrics 140(4), e20164102 (2017).
Størdal, K. et al. Review article: exposure to microbes and risk of coeliac disease. Aliment Pharmacol Ther 53(1), 43–62 (2021).
Goyal, D., Limesand, S. W. & Goyal, R. Epigenetic responses and the developmental origins of health and disease. J Endocrinol 242(1), T105–T119 (2019).
Lockett, G. A. et al. Association of season of birth with DNA methylation and allergic disease. Allergy 71(9), 1314–1324 (2016).
Jeppegaard, M., Rasmussen, S., Anhøj, J. & Winter, L. Winter, spring, summer or fall: temporal patterns in placenta-mediated pregnancy complications-an exploratory analysis. Arch Gynecol Obstet https://doi.org/10.1007/s00404-023-07094-6 (2023).
Dopico, X. C. et al. Widespread seasonal gene expression reveals annual differences in human immunity and physiology. Nat Commun 12(6), 7000 (2015).
Aronsson, C. A. et al. Age at Gluten Introduction and Risk of Celiac Disease. Pediatrics 135(2), 239–245 (2015).
Lindfors, K. et al. Metagenomics of the faecal virome indicate a cumulative effect of enterovirus and gluten amount on the risk of coeliac disease autoimmunity in genetically at risk children: The TEDDY study. Gut 69(8), 1416–1422 (2020).
Hummel, S. et al. Associations of breastfeeding with childhood autoimmunity, allergies, and overweight: The Environmental Determinants of Diabetes in the Young (TEDDY) study. Am J Clin Nutr 114(1), 134–142 (2021).
Stewart, C. J. et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562(7728), 583–588 (2018).
Davenport, E. R. et al. Seasonal variation in human gut microbiome composition. PLoS One 9(3), e90731 (2014).
Russell, J. T. et al. Genetic risk for autoimmunity is associated with distinct changes in the human gut microbiome. Nat Commun 10(1), 3621 (2019).
Acknowledgements
The TEDDY Study Group.
Funding
The study was supported specifically by the Foundation for Pediatric Research, the Competitive State Research Financing of the Expert Area of Tampere University Hospital, the Päivikki and Sakari Sohlberg Foundation. TEDDY is funded by U01 DK63829, U01 DK63861, U01 DK63821, U01 DK63865, U01 DK63863, U01 DK63836, U01 DK63790, UC4 DK63829, UC4 DK63861, UC4 DK63821, UC4 DK63865, UC4 DK63863, UC4 DK63836, UC4 DK95300, UC4 DK100238, UC4 DK106955, UC4 DK112243, UC4 DK117483, U01 DK124166, U01 DK128847, and Contract No. HHSN267200700014C from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institute of Environmental Health Sciences (NIEHS), Centers for Disease Control and Prevention (CDC), and Breakthrough T1D (formerly JDRF). A part of this work is supported by the NIH/NCATS Clinical and Translational Science Awards to the University of Florida (UL1 TR000064) and the University of Colorado (UL1 TR002535). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Consortia
Contributions
AE: Concept and design, statistical analysis and interpretation of data, drafting the manuscript, final approval of the version to be published; KLy: Concept and design, statistical analysis and interpretation of data, drafting the manuscript, critical revision of the manuscript for important intellectual content, final approval of the version to be published; KLi: Concept and design, interpretation of data, critical revision of the manuscript for important intellectual content, final approval of the version to be published; HP: Statistical analysis and interpretation of data, critical revision of the manuscript for important intellectual content, final approval of the version to be published; SK: Concept and design, critical revision of the manuscript for important intellectual content, final approval of the version to be published; EL: Concept and design, critical revision of the manuscript for important intellectual content, final approval of the version to be published; BA: Concept and design, critical revision of the manuscript for important intellectual content, final approval of the version to be published; WH: Concept and design, critical revision of the manuscript for important intellectual content, final approval of the version to be published; JK: Concept and design, critical revision of the manuscript for important intellectual content, obtaining funding, final approval of the version to be published; MR: Concept and design, critical revision of the manuscript for important intellectual content, final approval of the version to be published; JT: Concept and design, critical revision of the manuscript for important intellectual content, final approval of the version to be published; AZ: Concept and design, critical revision of the manuscript for important intellectual content, final approval of the version to be published; Agardh D: Concept and design, interpretation of data, drafting the manuscript, critical revision of the manuscript for important intellectual content, final approval of the version to be published; Kurppa K: Concept and design, interpretation of data, drafting the manuscript, critical revision of the manuscript for important intellectual content, final approval of the version to be published, study supervision. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Informed consent was obtained from all participating families. All research was performed in accordance with relevant regulations, and the study was performed in accordance with the Declaration of Helsinki. The study design and patient enrollment were approved by local U.S. Institutional Review Boards and European Ethics Committee Boards in Colorado’s Colorado Multiple Institutional Review Board 04–0361, Georgia’s Medical College of Georgia Human Assurance Committee (2004–2010), Georgia Health Sciences University Human Assurance Committee (2011–2012), Georgia Regents University Institutional Review Board (2013–2016), Augusta University Institutional Review Board (2017-present) HAC 0405380, Florida’s University of Florida Health Center Institutional Review Board IRB201600277, Washington state’s Washington State Institutional Review Board (2004–2012) and Western Institutional Review Board (2013–2019), WCG IRB (2020-present) 20130211, Finland’s Ethics Committee of the Hospital District of Southwest Finland Dnro168/2004, Germany’s Bayerischen Landesärztekammer (Bavarian Medical Association) Ethics Committee 04089, Sweden’s Regional Ethics Board in Lund, Sect. 2 (2004–2012) and Lund University Committee for Continuing Ethical Review (2013–2021), Swedish Ethical Review Authority (2022-present) 217/2004 and the study is monitored by External Advisory Board formed by the National Institutes of Health.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Eurén, A., Lynch, K., Lindfors, K. et al. Risk of celiac disease autoimmunity is modified by interactions between CD247 and environmental exposures. Sci Rep 14, 25463 (2024). https://doi.org/10.1038/s41598-024-75496-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-75496-w
