
Abstract
The strength of natural clay can be improved with epoxy resins. However, nanoscale curing mechanisms remain poorly understood, which is essential for enhancing stability. In this study, molecular dynamics simulation was employed to calculate the quantity of interface hydrogen bonds, adsorption energy, radius of gyration, and mechanical properties of clay cured by diglycidyl ether of bisphenol-A epoxy resin (DGEBA), diglycidyl ether 4,4’-dihydroxy diphenyl sulfone (DGEDDS), and Aliphatic epoxidation of olefin resin (AEOR). Adsorption behavior and mechanical properties of the clay cured by three epoxy resins were investigated: (1) The chain structure of AEOR led to 18.2% more hydrogen bonds than DGEBA and 59.1% more than DGEDDS. (2) The simulated adsorption energies for DGEBA, DGEDDS, and AEOR with kaolinite were 92.59, 98.25, and 116.87 kcal·mol−1, respectively. (3) The bulk and shear modulus of kaolinite increased by 4.93% and 4.80% when using AEOR. The interface stability and mechanical properties of kaolinite were also improved through strong hydrogen bonds and high adsorption energy. (4) The improvement in Young’s modulus of kaolinite was most significant with AEOR, followed by DGEDDS. AEOR excelled in the Z direction, while DGEDDS excelled in the X and Y directions. This research provided a theoretical foundation to effectively improve the properties of clay using epoxy resins.
Similar content being viewed by others


Introduction
Clay is widely utilized in road engineering1 and foundation construction2. Its loose structure leads to significant shrinkage and expansion, causing instability3. Traditional stabilization relies on mechanical compaction, which is noisy and slow4,5,6. In contrast, polymer-based chemical curing offers less curing time and enhanced stability7. Epoxy resins, essential thermosetting polymers in engineering, have found extensive applications in clay chemical curing8,9,10. Epoxy resins are low-molecular-weight prepolymers containing two or more epoxy groups11. Structurally, epoxy resins mainly include glycidyl ethers, glycidyl esters, glycidyl amines, aliphatic, and cycloaliphatic epoxy resins. Due to their excellent stability and curing properties, glycidyl ether epoxy resins like diglycidyl ether of bisphenol-A epoxy resins (DGEBA), diglycidyl ether 4,4’-dihydroxy diphenyl sulfone (DGEDDS), and Aliphatic epoxidation of olefin resin (AEOR) are often utilized as curing agents12,13,14,15. Most research on the curing of clay with polymers has primarily focused on experimental studies. The research on clay-polymer intercalation can be traced back to the 1980s16. Results in References17,18 indicate that laboratory-scale simulated aging tests show sand-fixing agent-poly and its composites enhance the compressive strength and wind erosion resistance of sand particles. The first study reporting the production of high-performance epoxy-clay nanocomposites was conducted by Kornmann et al.19. The results in References20,21 show that the macroscopic mechanical properties of epoxy-clay nanocomposites strongly depend on the type of nanocomposite structure. Current research has shown that most studies concentrate on macroscopic mechanical properties22. However, the nanoscale aspects of clay cured by epoxy resins are poorly studied, and the mechanisms remain unclear. The influence of nanoscale structure on adsorption behavior and mechanical properties is not well understood. Since the macroscopic properties of cured clay are largely determined by its nanoscale structure, investigating the nanoscale curing mechanisms of epoxy resins is crucial for their engineering applications.
Molecular dynamics simulation is one of the effective methods to reveal the nanoscale mechanisms of materials, offering a detailed perspective on atomic interactions23,24,25,26. It has been applied extensively in materials science to study the structural and mechanical properties of materials27,28. Molecular dynamics simulations have been utilized to quantitatively explore interactions between various materials at the nanoscale in previous studies10,29,30,31. These have provided theoretical foundations for understanding the binding mechanisms of surface-reactive groups, synthesis processes, and mechanical properties of materials. Therefore, employing molecular dynamics simulation to study the nanoscale mechanisms of clay cured by epoxy resins serves as a theoretical basis for determining the curing type and contents of curing agents.
Adsorption behavior is a crucial step in the process of clay cured by epoxy resins, determining the initial state of molecular motion within the process and serving as a key to understanding the curing mechanisms. Three essential parameters that delineate adsorption behavior are the quantity of interface hydrogen bonds, the adsorption energy and the radius of gyration. When in contact with soil particles, the polymer adheres to their surfaces due to electrostatic forces32, resulting in interparticle interactions, such as hydrogen bonds33, which substantially influence the effectiveness and performance of soil curing. Adsorption energy can be utilized to evaluate the strength and efficiency of interactions between various substances on mineral surfaces34. The radius of gyration is a parameter that describes the extent of a molecule’s expansion in space, providing insights into how the polymer adsorbs and unfolds on the soil surface35. In previous studies36,37,38, molecular dynamics simulation methods were employed to investigate the adsorption behavior between various organic materials and clay. These studies reveal that Interactions between substances are key factors affecting the stability and mechanical properties of clay composite materials. While the mentioned investigation on clay adsorption behavior mainly focuses on organic materials without epoxy groups, the nanoscale adsorption behavior in the field of clay cured by epoxy resins is still not understood. Therefore, conducting relevant research on the adsorption behavior of clay cured by epoxy resins is necessary.
Mechanical properties directly affect the stability and bearing capacity of clay and are crucial for assessing the effectiveness of clay curing. Mechanical parameters such as bulk modulus, shear modulus, and Young’s modulus characterize the response and deformation characteristics of soil structures under different types of stress and are key mechanical performance parameters to evaluate clay solidification effectiveness. Molecular dynamics simulation has been utilized to investigate the bulk modulus and shear modulus of clay composites under different environmental conditions in many previous studies39,40. However, due to differences in the study subjects, the obtained results cannot be directly applied to clay cured by epoxy resins. Moreover, the elastic constants of the nanoscale structure in clay cured by epoxy resins are lacking. Therefore, it is essential to analyze the mechanical properties of composite molecular systems comprising epoxy resins and clay to evaluate the curing effects of different epoxy resins.
Epoxy resins have been widely employed in curing clay. Relevant macroscopic and microscopic experiments have been investigated in previous studies41,42. However, the nanoscale curing mechanisms of clay from the atomic perspective are still not understood. Molecular models of epoxy resins and clay were constructed. Based on the optimized molecular models, the composite molecular systems between different types of epoxy resins and clay were constructed for molecular dynamics simulation. Through analyzing the quantity of interface hydrogen bonds and adsorption energy of composite molecular systems, a nanoscale perspective on the adsorption behavior between epoxy resins and clay was provided. Furthermore, calculations were conducted to analyze the mechanical properties of the composite molecular systems. This study aimed to construct molecular interface models of epoxy resins and clay and to investigate their curing mechanisms using molecular dynamics simulations. It provided essential insights for enhancing clay stability and performance, establishing a theoretical foundation for effective curing methods and optimal curing agent content.
Simulation method
Molecular models of epoxy resins
In this study, DGEBA, DGEDDS, and AEOR were utilized in the simulation. The molecular models of epoxy resins were constructed in Materials Studio software. Considering the practical engineering, the content of epoxy resin soil stabilizers is typically calculated by relative weight (the ratio of epoxy resin weight to clay weight)38. Therefore, the molecular weights of three epoxy resins are as close as possible while establishing the molecular models of epoxy resins. The parameters for the three epoxy resins are shown in Table 1. The structural formula is shown in Fig. 1a and molecular model of DGEBA is shown in Fig. 1b.

Structural formula and molecular model of DGEBA. (a) Structural formula; (b) molecular model.
DGEDDS is a common type of glycidyl ether-type epoxy resin42. The substitution of the isopropyl group in DGEBA with the strongly polar sulfonyl group leads to the formation of DGDDS. The structural formula is shown in Fig. 2a and molecular model of DGEDDS is shown in Fig. 2b.

Structural formula and molecular model of DGEBS. (a) Structural formula; (b) molecular model.
AEOR is characterized by a linear macromolecular structure, consisting of aliphatic chains in its molecular structure, devoid of benzene rings, cyclohexane rings, or heterocycles43. Its structure combines elements of both polybutadiene rubber and epoxy copolymer, resulting in its enhanced impact toughness and adhesive properties. The structural formula is shown in Fig. 3a and molecular model of AEOR is shown in Fig. 3b.

Structural formula and molecular model of AEOR. (a) Structural formula; (b) Molecular model.
Geometry optimization was applied to the initial structures to obtain the geometry optimized configuration, as reported in reference44. This was done to ensure that the initial structures of the simulation system were stable and reasonable.
Molecular model of clay
In this study, kaolinite was utilized in the simulation, primarily composed of kaolinite minerals with the molecular formula [Si2O4][Al2O(OH)4]. It is a layered silicate clay mineral, constituting approximately 10 – 95% of kaolinite minerals45,46. Previous study has shown that a single-layer kaolinite crystal structure model effectively represents the interaction between the kaolinite surface and organic molecules33. Thus, a single-layer kaolinite crystal structure model was utilized in this study to construct the interface models between kaolinite surface and epoxy resins. The crystal structure model of the single-layer kaolinite (0 0 1) is shown in Fig. 4.

Crystal structure model of single-layer kaolinite (0 0 1).
Based on the kaolinite unit cell model, a supercell model was constructed, and its (0 0 1) surface was cleaved, followed by the creation of a vacuum slab with a thickness of 30 Å. The total atoms of kaolinite were 2040. Additionally, to protect the model structure against boundary conditions effects, periodic boundary conditions were utilized. This was done to ensure that interactions between molecules within the system were not affected by boundary molecules, thus preventing any unreasonable simulation results. The crystal structure molecular model of kaolinite is shown in Fig. 5.

Crystal structure model of kaolinite.
Based on the models of the three epoxy resins and kaolinite that have been constructed, interface models between epoxy resins and kaolinite were established. The interface model formed between DGEBA and kaolinite was referred to as DGEBA-Kaolinite. Similarly, the interface model formed between DGEDDS or AEOR and kaolinite was referred to as DGEDDS-Kaolinite or AEOR-Kaolinite, respectively. Three interface models are shown in Fig. 6.

Interface models of epoxy resin and kaolinite. (a) DGEBA-Kaolinite, (b) DGEDDS-Kaolinite, (c) AEOR-Kaolinite.
Simulation details
The molecular models of epoxy resins and kaolinite were constructed in Material Studio software. The simulation was carried out employing the Universal Force Field (UFF), a commonly employed classical force field within molecular dynamics simulations, relying on empirical potential functions for atomic interactions47. UFF is usually applied to describe atomic interactions in molecules and encompasses parameters for almost all elements in the periodic table. UFF is known for its versatility, as it can be effectively utilized for various organic and inorganic compounds35. Furthermore, it has the capability to predict the physical properties and chemical reactions of molecules based on their geometric structure and elemental composition48.
In this study, the canonical NVT ensemble was employed with 298 K temperature, and the total simulation duration lasted for 10 ns. The initial velocities of system atoms were randomly assigned in accordance with the Maxwell–Boltzmann distribution at the initial temperature. When the total energy of system converged, the system reached an equilibrium state. To prevent being confined within a singular potential well, three different epoxy resins were placed at least 5 Å above the initial kaolinite molecular model. At this position, the analysis of adsorption energies and interface hydrogen bonds for all configurations utilized the default values of the software (2.5 Å as the maximum hydrogen-receptor distance and 90° as the minimum donor-hydrogen-acceptor angle). The flowchart of main algorithm for simulation was shown in Fig. 7.

Flowchart of main algorithm of simulation.
Results and discussion
Adsorption behavior analysis
Hydrogen bonds
The hydrogen bond is a special type of chemical bond that constitutes weak interaction between certain molecules, playing a significant role in adsorption processes. The quantity of hydrogen bonds formed between epoxy resins and the surface of kaolinite was calculated, as shown in Fig. 8.

Hydrogen bonds formed between epoxy resin and kaolinite surface (Hydrogen bonds are shown as light blue dashed lines). (a) DGEBA-Kaolinite; (b) DGEDDS-Kaolinite; (c) AEOR-Kaolinite.
The results indicated that the quantity of hydrogen bonds within the lowest energy conformations of DGEBA-Kaolinite, DGEDDS-Kaolinite, and AEOR-Kaolinite was 7, 5, and 9, respectively. The quantity of interface hydrogen bonds formation was influenced by the reactant content. Therefore, to provide a more reasonable comparison of the adsorption efficiency of the three epoxy resins on kaolinite mineral surfaces, the concept of hydrogen bonds per unit molecular weight of the epoxy resin(\(N_{{{\text{unit}}}}\)) was calculated by Eq. (1), as reported in reference38:
where \(N_{{{\text{surface}}}}\) is the quantity of hydrogen bonds generated on kaolinite surface; \(m\) is molecular weight of the epoxy resin. The hydrogen bonds of DGEBA-Kaolinite, DGEDDS-Kaolinite, and AEOR-Kaolinite at unit molecular weight of epoxy resin are displayed in Table 2.
The results showed that per molecular weight of AEOR formed the highest quantity among the three epoxy resins, surpassing DGEBA and DGEDDS by 18.2% and 59.1%, respectively. A higher quantity of hydrogen bonds contributed to the enhanced stability of the clay. DGEBA, DGEDDS, and AEOR molecules formed hydrogen bonds with the hydrogen atoms on the surface of kaolinite through their oxygen atoms. The hydrogen bond locations were concentrated at the terminal groups of DGEBA, DGEDDS, and AEOR, which typically contained hydroxyl (-OH) or ether (-O-) groups. Additionally, the oxygen atoms in the sulfonyl group (O = S = O) of DGEDDS, due to their high electronegativity, were prone to form strong hydrogen bonds with the hydrogen atoms on the surface of kaolinite. The chain-like structure of AEOR allowed it to cover a larger surface area and form more hydrogen bonds. Compared to rigid or cyclic molecules, chain-like AEOR were more easily conformed on the kaolinite surface, thereby increasing the area of their interaction with its surface. The flexibility of the molecular chains implied that AEOR molecules could better adjust themselves, allowing multiple functional groups to come closer to the hydrogen-bond-forming sites on the kaolinite surface, thereby increasing the number of hydrogen bonds.
Adsorption energy
In molecular dynamics simulation, interaction energy is a crucial physical quantity utilized to describe the binding strength between interacting molecules. Therefore, it could be utilized to assess the intensity of intermolecular interaction forces and the stability of composites34. Since the interaction energy in complexes tends to bring molecules closer together, the formation of complexes typically results in the release of energy, making the interaction energy a negative value. The reciprocal of interaction energy is referred to as adsorption energy, which is a relative measure utilized to compare the stability and strength of interactions between different systems, helping to study the effects of intermolecular interactions35,38. A larger adsorption energy indicates a stronger interaction and greater affinity between epoxy resins and kaolinite. In molecular dynamics simulations, force fields are commonly utilized to describe intermolecular interactions, which include bond energy terms, bond angle energy terms, and non-bonded interaction terms. The adsorption energies were calculated by Eq. (2), as reported in reference35:
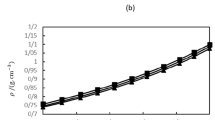
where \(E_{{{\text{total}}}}\) represents the total energy of the entire system, containing all atoms, in \({\text{kcal}} \cdot {\text{mol}}^{ - 1}\);\(E_{{{\text{epoxy}}}}\) represents the configuration energy of epoxy resin molecules, in \({\text{kcal}} \cdot {\text{mol}}^{ - 1}\); \(E_{{{\text{kao}}}}\) is the energy associated with the kaolinite, in \({\text{kcal}} \cdot {\text{mol}}^{ - 1}\); \(E_{{{\text{ads}}}}\) represents the adsorption energy between epoxy resin and kaolinite, in \({\text{kcal}} \cdot {\text{mol}}^{ - 1}\), which dictates the energy necessary to disrupt the epoxy resin and kaolinite interaction. At the same time, the value also reflects the strength of adhesion at the interface and characterizes the adhesive strength between epoxy resins and kaolinite38. Figure 9 shows the variation curves of adsorption energies for the three interface models of epoxy resins and kaolinite, while Fig. 10 presents the corresponding adsorption energies.

Energies change of three interface models in 10 ns.

Adsorption energies of the three epoxy resins and kaolinite.
The adsorption energies of three interface models of epoxy resins and kaolinite stabilized within 4.0 ns and were in a fluctuating state around equilibrium. AEOR showed the largest fluctuations in energy compared to DGEBA and DGEDDS. This could indicate more dynamic interactions on the kaolinite surface, due to the flexibility of AEOR’s chain structure, allowing it to adapt more dynamically to the kaolinite surface. The results of adsorption energies for DGEBA, DGEDDS, and AEOR with kaolinite were 92.59, 98.25, and 116.87 \({\text{kcal}} \cdot {\text{mol}}^{ - 1}\), respectively. The results indicated that AEOR exhibited the highest adsorption energy with kaolinite. This suggested the strongest affinity between AEOR and kaolinite among the three epoxy resins, which was consistent with the hydrogen bond formation results.
Although DGEBA formed more hydrogen bonds with the kaolinite surface than DGEDDS, DGEDDS exhibited greater adsorption energy. This is due to the higher electronegativity of the oxygen atoms in the sulfonyl group (O = S = O) of DGEDDS. These highly polar sulfonyl groups could interact strongly with the kaolinite surface through electrostatics. The relatively high surface energy of DGEDDS indicates a strong interaction force with kaolinite, resulting in stronger hydrogen bonds with the kaolinite surface hydrogen atoms and increased adsorption energy. This enhanced intermolecular interaction makes DGEDDS more stable on the surface.
Radius of gyration
The spatial dimensions of a molecule could be defined through the parameter known as its radius of gyration (\(R_{g}\)), which was calculated by Eq. (3). The \(R_{g}\) of DGEBA, DGEDDS, and AEOR on the kaolinite surface in the lowest energy conformations are shown in Fig. 11.
where \(m_{i}\) is the mass of the atom \(i\), \(r_{i}\) is considered alongside the position of the atom \(i\), in relation to the center of mass of the molecule.

The radius of gyration of DGEBA,DGEDDS and AEOR at the kaolinite surface. (a) DGEBA; (b) DGEDDS; (c) AEOR.
At the beginning of the simulation, DGEBA approached and adsorbed onto the surface of kaolinite rapidly. This caused the conformation to quickly transition from an extended state to a compact state, resulting in the \(R_{g}\) decreasing sharply from nearly 1.30 to 0.70 nm. After approximately 2 ns, the \(R_{g}\) gradually stabilized, with the amplitude of fluctuation decreasing and remaining between 0.50 and 0.60 nm. This stable state indicated that DGEBA had predominantly adsorbed onto the kaolinite surface and formed a relatively stable conformation.
DGEDDS rapidly approached and partially adsorbed onto the kaolinite surface within the first 1 ns of the simulation, forming a more compact conformation. Consequently, the \(R_{g}\) decreased sharply within the first 1 ns. After 2 ns, the adsorption state of DGEDDS on the kaolinite surface tended to stabilize, with the \(R_{g}\) remaining in the range of 1.03 to 1.08 nm. Compared to DGEBA, the stabilized \(R_{g}\) for DGEDDS on the kaolinite surface was larger, indicating that its adsorption efficiency was slightly lower than that of DGEBA.
Compared to DGEBA and DGEDDS, AEOR, due to its chain-like structure, exhibited higher conformational flexibility and a larger contact area. This characteristic allowed AEOR to maintain a certain degree of conformational freedom and undergo frequent conformational changes during the adsorption process with kaolinite, resulting in significant fluctuations in the \(R_{g}\).
Mechanical properties analysis
Mechanical property simulations and analysis were performed on the optimized configurations. Under constant temperature \(T\), the relationship between the elastic stiffness coefficient \(C_{ijkl}\), stress \(\sigma_{ij}\), and strain \(\varepsilon_{kl}\) could be described by Eq. (4), as reported in reference35:
where \(V_{0}\) represents the volume of the simulation cell the undeformed configuration, in \({\text{m}}^{{3}}\), and \(A\) represents the Helmholtz free energy, in \(J\).
Based on the elastic tensor, the Voigt -Reuss-Hill (VRH) approximation40,49 was utilized to calculate the bulk modulus and shear modulus of the systems in this study. Assuming each component has the same strain as the polycrystal, Voigt50 denotes the polycrystal’s bulk modulus (\(K_{V}\)) and shear modulus (\(G_{V}\)) as Eqs. (5) and (6). Reuss51, on the other hand, assumes the same stress and denotes the polycrystal’s bulk modulus (\(K_{R}\)) and shear modulus (\(G_{R}\)) as Eqs. (7) and (8). Hill49 indicates that the Voigt and Reuss models can serve as the respective upper and lower bounds for the average elastic modulus in polycrystalline materials. Consequently, the Eqs. (9) and (10) define the bulk modulus (\(K\)) and shear modulus (\(G\)) as the arithmetic averages of the Voigt and Reuss models.
where subscripts \(V\) and \(R\) are the Reuss and Voigt averages, respectively. \(S_{ij}\) is the elastic compliance matrix components obtained from the inverse of the elastic stiffness matrix, in \(m^{2} \cdot N\).
The calculated results of the bulk modulus and shear modulus of different interface models are shown in Table 3. The mechanical properties of clay have been investigated in previous studies52,53. The simulated bulk modulus in the previous study ranges from 38.00 to 80.00 GPa under different conditions52, and the bulk modulus in this study fell within the range of 47.00 to 50.00 GPa, showing good agreement with the results of the previous study. Furthermore, the simulated bulk modulus of kaolinite in this study was 47.42 GPa, which was consistent with the experimental bulk modulus value of well-crystallized kaolinite reported in previous studies, which is 47.90 ± 8.00 GPa53. The values of the shear modulus obtained in this study are close to those in previous study52, which ranges from 18.00 to 42.00 GPa.
The results of the Hill values of bulk modulus and shear modulus demonstrated that the addition of all three epoxy resins increased the compressive and shear deformation resistance of kaolinite. Consistent with the results of hydrogen bonds and adsorption energy, the bulk modulus of AEOR-Kaolinite was the highest among the four systems, being 4.93% higher than that of kaolinite. At the same time, the shear modulus of AEOR-Kaolinite was the highest among the four systems, and it was 4.80% higher than that of kaolinite. Hydrogen bonds are able to enhance the bonding force between epoxy resin and kaolinite, making the interface more stable. High adsorption energy indicates strong adsorption and interaction between the epoxy resin and kaolinite, which contributes to the improvement of the interface’s stability and mechanical properties.
Young’s modulus (\(E\)) is one of the important parameters describing the mechanical properties of materials and is utilized to assess the stiffness of materials for linear elastic deformation when subjected to a force. Based on the calculations of bulk modulus (\(K\)) and shear modulus (\(G\)) as obtained above, Young’s modulus (\(E\)) can be defined by Eq. (11), as reported in reference49. The calculated results of the Young’s modulus of different interface models are shown in Table 4.
The results indicated that three epoxy resins all effectively improved the Young’s modulus of kaolinite. Among the epoxy resins, DGEBA exhibited relatively weaker bonding with kaolinite. However, due to the structural characteristics and molecular arrangement of the interface in the Z-direction, a significant enhancement in the Young’s modulus in the Z-direction was still observed. DGEDDS, with its greater number of hydrogen bonds and moderate adsorption energy, showed stronger interface bonding, leading to improvements in Young’s modulus in the X and Y directions, while the increase in the Z-direction was slightly smaller compared to DGEBA. AEOR, due to its chain-like structure and the highest number of hydrogen bonds formed, demonstrated the strongest interface bonding and the highest adsorption energy. This resulted in a significant increase in the Young’s modulus in all directions, with the greatest enhancement observed in the Z-direction, which was related to its strong interface bonding and favorable molecular arrangement. Consistent with the trend in Young’s modulus improvement, the higher bulk modulus and shear modulus reflected the enhanced overall rigidity and resistance to deformation of the material after adsorption. In terms of mechanical properties, AEOR exhibited the most significant improvement among the three epoxy resins.
Conclusions
This study investigated the nanoscale models of DGEBA-Kaolinite, DGEDDS-Kaolinite, and AEOR-Kaolinite through molecular dynamics simulation, aiming to elucidate the curing mechanisms of kaolinite cured by epoxy resins. The primary findings were summarized as follows.
-
1.
Compared to DGEBA and DGEDDS, AEOR, due to its chain-like structure, exhibited higher conformational flexibility and a larger contact area, resulting in significant fluctuations in the radius of gyration. This characteristic allowed AEOR to maintain a certain degree of conformational freedom, undergo frequent conformational changes, and form more hydrogen bonds on the surface of kaolinite.
-
2.
AEOR had the highest adsorption energy on the surface of kaolinite among the three epoxy resins, including DGEBA, DGEDDS and AEOR, indicating the strongest affinity. DGEDDS showed higher electronegativity in its sulfonyl group oxygen atoms, leading to the formation of stronger hydrogen bonds with kaolinite, thereby increasing adsorption energy.
-
3.
The three epoxy resins enhanced the bulk and shear modulus of kaolinite, with AEOR showing the most significant improvement in both. Hydrogen bonds strengthen the bonding force between epoxy resin and kaolinite, stabilizing the interface. High adsorption energy indicates strong interaction, improving interface stability and mechanical properties.
-
4.
The three epoxy resins improved Young’s modulus of kaolinite. DGEDDS had more hydrogen bonds and moderate adsorption energy, enhancing Young’s modulus in X and Y directions more than in the Z direction compared to DGEBA. AEOR, with its chain-like structure and the most hydrogen bonds, resulted in the strongest interface bonding and highest adsorption energy, increasing Young’s modulus in all directions, especially in the Z direction. Higher bulk modulus and shear modulus indicated increased rigidity and resistance to deformation after adsorption. AEOR had the most significant improvement in mechanical properties among the resins.
Data availability
Data is provided within the manuscript.
References
Elhassan, A. A. M. et al. Effect of clay mineral content on soil strength parameters. Alex. Eng. J.63, 475–485 (2023).
Sun, Y. Y. & Xiao, H. J. Wall displacement and ground-surface settlement caused by pit-in-pit foundation pit in soft clays. KSCE J. Civ. Eng.25(4), 1262–1275 (2021).
Miao, F. S., Wu, Y. P., Török, Á., Li, L. W. & Xue, Y. Centrifugal model test on a riverine landslide in the Three Gorges Reservoir induced by rainfall and water level fluctuation. Geosci. Front.13(3), 101378 (2022).
Gillott, J. Some clay-related problems in engineering geology in North America. Clay Miner.21(3), 261–278 (1986).
Mohamed, A. E. M. K. Improvement of swelling clay properties using hay fibers. Constr. Build. Mater.38, 242–247 (2013).
Afrin, H. A review on different types soil stabilization techniques. Int. J. Transp. Eng. Technol.3(2), 19–24 (2017).
Zandieh, A. R. & Yasrobi, S. S. Retracted article: Study of factors affecting the compressive strength of sandy soil stabilized with polymer. Geotech. Geol. Eng.28, 139–145 (2010).
Huang, W., Liu, Z., Zhou, C. Y. & Yang, X. Enhancement of soil ecological self-repair using a polymer composite material. Catena188, 104443 (2020).
Ellis, B. Chemistry and Technology of Epoxy Resins (Springer, 1993).
Zhang, Y. C., Liu, X. D., Zhang, C. & Lu, X. C. A combined first principles and classical molecular dynamics study of clay-soil organic matters (SOMs) interactions. Geochimica et Cosmochimica Acta291, 110–125 (2020).
Jin, F. L., Li, X. & Park, S. J. Synthesis and application of epoxy resins: A review. J. Ind. Eng. Chem.29, 1–11 (2015).
Shi, Y. F., Sun, Y. Y., Gao, B., Xu, H. X. & Wu, J. C. Importance of organic matter to the retention and transport of bisphenol A and bisphenol S in saturated soils. Water Air Soil Pollut.230, 1–9 (2019).
Jiang, T. W. et al. Recycling waste polycarbonate to bisphenol A-based oligoesters as epoxy-curing agents, and degrading epoxy thermosets and carbon fiber composites into useful chemicals. ACS Sustain. Chem. Eng.10(7), 2429–2440 (2022).
Dagdag, O. et al. Anticorrosive performance of new epoxy-amine coatings based on zinc phosphate tetrahydrate as a nontoxic pigment for carbon steel in NaCl medium. Arab. J. Sci. Eng.43, 5977–5987 (2018).
Park, S. S., Lee, J. S., Yoon, K. B., Woo, S. W. & Lee, D. E. Application of an acrylic polymer and epoxy emulsion to red clay and sand. Polymers13(19), 3410 (2021).
Gao, F. Clay/polymer composites: The story. Mater. Today7(11), 50–55 (2004).
Yang, J., Wang, F., Fang, L. & Tan, T. Synthesis, characterization and application of a novel chemical sand-fixing agent-poly (aspartic acid) and its composites. Environ. Pollut.149(1), 125–130 (2007).
Yang, J., Wang, F., Fang, L. & Tan, T. The effects of aging tests on a novel chemical sand-fixing agent—Polyaspartic acid. Compos. Sci. Technol.67(10), 2160–2164 (2007).
Kornmann, X., Berglund, L. A., Thomann, R., Mulhaupt, R. & Finter, J. High performance epoxy-layered silicate nanocomposites. Polym. Eng. Sci.42(9), 1815–1826 (2002).
Zabihi, O., Ahmadi, M., Nikafshar, S., Preyeswary, K. C. & Naebe, M. A technical review on epoxy-clay nanocomposites: Structure, properties, and their applications in fiber reinforced composites. Compos. Part B Eng.135, 1–24 (2018).
Moghadam, R. M., Saber-Samandari, S. & Hosseini, S. A. On the tensile behavior of clay–epoxy nanocomposite considering interphase debonding damage via mixed-mode cohesive zone material. Compos. Part B Eng.89, 303–315 (2016).
Marto, A., Latifi, N. & Sohaei, H. Stabilization of laterite soil using GKS soil stabilizer. Electron. J. Geotech. Eng.18(18), 521–532 (2013).
Zhu, J., Shen, D. J., Jin, B. S. & Wu, S. X. Theoretical investigation on the formation mechanism of carbonate ion in microbial self-healing concrete: Combined QC calculation and MD simulation. Constr. Build. Mater.342, 128000 (2022).
Zhu, J., Shen, D. J., Wu, W., Jin, B. S. & Wu, S. X. Hydration inhibition mechanism of gypsum on tricalcium aluminate from ReaxFF molecular dynamics simulation and quantum chemical calculation. Mol. Simul.47(17), 1465–1476 (2021).
Zhu, J., Shen, D. J., Xie, J. J., Jin, B. S. & Wu, S. X. Transformation mechanism of carbamic acid elimination and hydrolysis reaction in microbial self-healing concrete. Mol. Simul.48(8), 719–735 (2022).
Zhu, J. et al. Mechanism of urea decomposition catalyzed by Sporosarcina pasteurii urease based on quantum chemical calculations. Mol. Simul.47(16), 1335–1348 (2021).
Singh, S. K., Chaurasia, A. & Verma, A. Basics of density functional theory, molecular dynamics, and monte carlo simulation techniques in materials science. Coating materials: Computational aspects, applications and challenges. Springer, pp. 111–124 (2023).
Kumar, G., Mishra, R. R., & Verma, A. Introduction to molecular dynamics simulations. Forcefields for atomistic-scale simulations: materials and applications, Springer, pp. 1–19 (2022).
Yuan, Y., Zhan, W. Q., Yi, H., Zhao, Y. L. & Song, S. X. Molecular dynamics simulations study for the effect of cations hydration on the surface tension of the electrolyte solutions. Colloids Surf. A Physicochem. Eng. Asp.539, 80–84 (2018).
Wang, H., Zhang, H., Liu, C. B. & Yuan, S. L. Coarse-grained molecular dynamics simulation of self-assembly of polyacrylamide and sodium dodecylsulfate in aqueous solution. J. Colloid Interface Sci.386(1), 205–211 (2012).
Prasitnok, O. & Prasitnok, K. Molecular dynamics simulations of copolymer compatibilizers for polylactide/poly (butylene succinate) blends. Phys. Chem. Chem. Phys.25(7), 5619–5626 (2023).
Song, Z. Z. et al. Laboratory and field experiments on the effect of vinyl acetate polymer-reinforced soil. Appl. Sci.9(1), 208 (2019).
Murgich, J., Rodríguez, J., Izquierdo, M. A., Carbognani, L. & Rogel, E. Interatomic interactions in the adsorption of asphaltenes and resins on kaolinite calculated by molecular dynamics. Energy Fuels12(2), 339–343 (1998).
Rai, B., Sathish, P., Tanwar, J., Moon, K. & Fuerstenau, D. A molecular dynamics study of the interaction of oleate and dodecylammonium chloride surfactants with complex aluminosilicate minerals. J. Colloid Interface Sci.362(2), 510–516 (2011).
Yan, L. J., Yang, Y., Jiang, H., Zhang, B. J. & Zhang, H. The adsorption of methyl methacrylate and vinyl acetate polymers on α-quartz surface: A molecular dynamics study. Chem. Phys. Lett.643, 1–5 (2016).
Wu, M. et al. Adsorption behaviour and mechanism of benzene, toluene and m-xylene (BTX) solution onto kaolinite: Experimental and molecular dynamics simulation studies. Sep. Purif. Technol.291, 120940 (2022).
Sikdar, D., Katti, D. R. & Katti, K. S. The role of interfacial interactions on the crystallinity and nanomechanical properties of clay–polymer nanocomposites: A molecular dynamics study. J. Appl. Polym. Sci.107(5), 3137–3148 (2008).
Huang, W., Geng, X. Y., Liu, Z. & Zhou, C. Y. Molecular dynamics study of polymeric stabilizers as soil improvement materials. Chem. Phys. Lett.806, 139985 (2022).
Anoukou, K., Zaoui, A., Zaïri, F., Naït-Abdelaziz, M. & Gloaguen, J. M. Molecular dynamics study of the polymer clay nanocomposites (PCNs): Elastic constants and basal spacing predictions. Comput. Mater. Sci.77, 417–423 (2013).
Du, J. P. et al. Modeling microstructural mechanical behavior of expansive soil at various water contents and dry densities by molecular dynamics simulation. Comput. Geotech.158, 105371 (2023).
Lim, S. J. & Kim, D. S. Effect of functionality and content of epoxidized soybean oil on the physical properties of a modified diglycidyl ether of bisphenol A resin system. J. Appl. Polym. Sci.138(20), 50441 (2021).
Adibzadeh, E., Mirabedini, S. M., Behzadnasab, M. & Farnood, R. R. A novel two-component self-healing coating comprising vinyl ester resin-filled microcapsules with prolonged anticorrosion performance. Prog. Org. Coat.154, 106220 (2021).
Varganici, C. D. et al. Effect of hardener type on the photochemical and antifungal performance of epoxy and oligophosphonate S-IPNs. Polymers14(18), 3784 (2022).
Schlegel, H. B. Geometry optimization. Wiley Interdiscip. Rev. Comput. Mol. Sci.1(5), 790–809 (2011).
Alaba, P. A., Sani, Y. M. & Daud, W. M. A. W. Kaolinite properties and advances for solid acid and basic catalyst synthesis. RSC Adv.5(122), 101127–101147 (2015).
Bish, D. L. Rietveld refinement of the kaolinite structure at 15 K. Clays Clay Minerals41, 738–744 (1993).
Rappé, A. K., Casewit, C. J., Colwell, K., Goddard, W. A. III. & Skiff, W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc.114(25), 10024–10035 (1992).
Sun, H. Ab initio characterizations of molecular structures, conformation energies, and hydrogen-bonding properties for polyurethane hard segments. Macromolecules26(22), 5924–5936 (1993).
Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. Sect. A65(5), 349 (1952).
Voigt, W. Lehrbuch der kristallphysik (Teubner, 1928).
Reuß, A. Berechnung der fließgrenze von mischkristallen auf grund der plastizitätsbedingung für einkristalle. ZAMM J. Appl. Math. Mech.9(1), 49–58 (1929).
Carrier, B., Vandamme, M., Pellenq, R. J. & Van Damme, M. H. Elastic properties of swelling clay particles at finite temperature upon hydration. J. Phys. Chem. C118(17), 8933–8943 (2014).
Wang, Z. J., Wang, H. & Cates, M. E. Effective elastic properties of solid clays. Geophysics66(2), 428–440 (2001).
Acknowledgements
The authors gratefully acknowledge the financial support of the Fundamental Research Funds for Central Universities of China (Grant No. B230201060) and JSTI Group (Grant NO. 823088816).
Author information
Authors and Affiliations
Contributions
D.J.S. conceptualization, funding acquisition; S.J.T writing–original draft, methodology; X.W. writing–review & editing, investigation; L.L.C. project administration; C.Y.W. visualization; R.X.L. visualization. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tao, S., Shen, D., Wang, X. et al. Molecular dynamics investigation of epoxy resin adsorption mechanisms on clay surfaces and the mechanical properties of epoxy resin-clay. Sci Rep 14, 26372 (2024). https://doi.org/10.1038/s41598-024-76950-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-76950-5
