
Abstract
Subarachnoid hemorrhage (SAH) is the deadliest form of hemorrhagic stroke; however, effective therapies are still lacking. Perfluorocarbons (PFCs) are lipid emulsion particles with great flexibility and their much smaller size as compared to red blood cells (RBCs) allows them to flow more efficiently within the blood circulation. Due to their ability to carry oxygen, a specific PFC-based emulsion, PFC-Oxygent, has been used as a blood substitute; however, its role in cerebral blood flow regulation is unknown. Adult C57BL/6 wildtype male mice were subjected to an endovascular perforation model of SAH followed by an intravenous (i.v.) injection of 9 ml/kg PFC-Oxygent or no treatment at 5 h after SAH. At 48 h after SAH, functional and anatomical outcomes were assessed. We found that SAH resulted in significant neurologic and motor deficits which were prevented by PFC-Oxygent treatment. We found that SAH-induced vasospasm, reduced RBC deformability, and augmented endothelial dysfunction were also restricted by PFC-Oxygent treatment. Moreover, mitochondrial activity and fusion proteins were also markedly decreased as assessed by oxidative phosphorylation (OXPHOS) after SAH. Interestingly, PFC-Oxygent treatment brought the mitochondrial activity close to the basal level. Moreover, SAH attenuated the level of phosphorylated AMP-activated protein kinase (pAMPK), whereas PFC treatment improved pAMPK levels. These data show the beneficial effects of PFC-Oxygent in limiting the severity of SAH. Further studies are needed to fully understand the mechanism through which PFC-Oxygent exerts its beneficial effects in limiting SAH severity.
Similar content being viewed by others


Introduction
Subarachnoid hemorrhage (SAH) is reported to be the most devastating form of hemorrhagic stroke leading to significant morbidity and mortality1. Immediately following SAH, there is an acute increase in intracranial pressure and a reduction in cerebral blood flow (CBF)2. These incidences lead to an early brain injury (EBI) and/or vasospasm-mediated delayed cerebral ischemia (DCI). Early brain injury is reported to develop within the first 72 h after SAH, whereas DCI appears to develop around day 7 in humans and within 24–48 h in rodents. In patients, EBI is reported to be a major factor leading to poor outcomes after SAH3,4. Recent pieces of evidence implicate EBI in the onset of vasospasm thereby leading to DCI. Various clinical and preclinical studies suggest that vasospasm gradually restricts blood flow to the affected regions of the brain5. With a decrease in CBF, there is also a gradual decrease in tissue oxygenation eventually leading to DCI5. DCI is reported to inflict severe neurologic deficits or even death in approximately half of the patients suffering from SAH6.
AMP-activated protein kinase (AMPK) enzyme plays a critical role in regulating cellular energy metabolism7. It is activated when the ratio of adenosine triphosphate (ATP) to adenosine monophosphate (AMP) decreases, which occurs when the cell is low on energy8. When AMPK is activated, it promotes energy-producing processes, such as glucose uptake and fatty acid oxidation, while inhibiting energy-consuming processes, such as protein synthesis and fatty acid synthesis8. This helps the cell adapt to a low-energy state and maintain energy homeostasis. It has been reported that oxygen is required to maintain normal energy metabolism through AMPK activation. Recent reports suggest that activation of AMPK may have a protective effect on the brain after SAH by reducing oxidative stress and inflammation and improving cellular energy metabolism9,10. Mitochondria are dynamic organelles that constantly undergo fission and fusion to maintain cellular metabolism8. After SAH, there is a surge in mitochondrial dysfunction leading to oxidative stress which subsequently results in neural cell death and neurologic impairments11. Moreover, mitochondrial reactive oxygen species (ROS) also play a significant role in the regulation of metabolic and mitochondrial functions through regulating AMPK activity12,13,14.
PFCs are fluorinated, inert organic compounds coated with an emulsifier, and a stabilizer that dissolves large volumes (50-fold that of plasma) of respiratory gases including O2. Due to their unique enhanced solubility for O2, PFCs have been used for years in research as enhanced gas transporter medium15. The special biological and chemical characteristics of PFCs allow their moderate dosages to be biocompatible16,17. Based on their small median size of ~ 0.2 μm and ability to carry O2, PFC-based emulsions have been investigated for their role as oxygen therapeutics in traumatic brain injury (TBI), decompression illness, heart attack, and ischemia18,19. A specific PFC, PFC-Oxygent, is reported to have a long half-life and has been used as oxygen therapeutic in an animal model of TBI19. In the current study, we tested the efficacy of PFC-Oxygent (hereafter referred to as PFC) in reducing the severity of SAH in an endoperforation model in mice.
Materials and methods
Animals and experimental groups
Male C57BL/6 wildtype (WT) mice of 8–10 weeks of age were maintained and housed in the animal facility at Henry Ford Health. All the experiments were conducted according to the protocol approved by the Henry Ford Health Institutional Animal Care and Use Committee (IACUC). This study was conducted following the ARRIVE guidelines. All experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Mice were randomly divided into three groups, Sham (n = 10), SAH (n = 13), and SAH with PFC (SAH + PFC, n = 13). Three mice from the SAH group and 2 mice from the SAH + PFC group died within 24 h after SAH. One mouse from the SAH + PFC group was excluded due to incomplete perforation as observed by no change in CBF immediately after perforation. Mice were kept on a 12-hour dark/light cycle and were provided with food and water ad libitum.
Randomization, Exclusion, and blinding
Randomization was performed by a person who was not involved with experimental design and group allocations. The mice that died or were excluded from the study based on our preset exclusion criteria were not included in the study. The personnel performing the surgeries, neurobehavioral testing, and all assays were blinded to the experimental groups. All outcomes were quantified in a blinded manner.
Induction of subarachnoid hemorrhage and PFC treatment
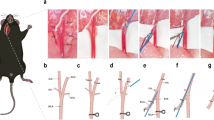
Mice were subjected to SAH, or sham surgery as previously described by us20. Briefly, mice were anesthetized with 3% isoflurane and maintained with 1.5 -2% isoflurane using SomnoSuite Low-Flow Anesthesia System (Kent Scientific Corp., Torrington, CT). A 5 − 0 size nylon monofilament was advanced into the ICA towards the circle of Willis until a slight resistance was felt. At that point, the filament was advanced further to perforate the blood vessel along the circle of Willis. Then the filament was immediately retracted, ECA was ligated, and the incision was sutured. Sham control mice underwent the same surgical procedure without the vessel perforation. Once the mice recovered from anesthesia and exhibited ambulatory movements, they were transferred to their original cages and returned to the animal facility. At 5 h after SAH, mice were treated with a single dose of 9 ml/kg PFC intravenously (i.v.) via the tail vein. Sham surgery mice did not receive any treatment. PFC-Oxygent was generously provided by Professor Bruce Spiess, Vice Chair of Research, Department of Anesthesiology, University of Florida.
Behavioral assessment
All mice were given training before SAH induction to familiarize themselves with the behavioral arena. At 48 h after SAH, mice were subjected to the following functional assessments.
Neurological deficit score (NDS)
Mice were tested for neurological scores as previously described20,21. The animals were scored for gait, body symmetry, circling, climbing, front limb symmetry, and compulsory circling. Each of these tests was graded from 0 to 4, thus establishing a maximum score of 24 points.
Rotarod test
Motor coordination was assessed by using the rotarod test as reported by us20. The mice were placed on a rotating beam that started at 5 rotations per minute (rpm) and gradually accelerated at a fixed interval to reach a maximum speed of 30 rpm. Each trial had a maximum duration of 5 min. Each group of mice underwent three trials at a 30-minute interval. The total time of retention on the rod for each mouse was automatically recorded and reported as latency to fall.
Hanging wire test
Front limb strength was assessed using the hanging wire test as reported by us22. A wire was stretched between two poles 100 cm apart and 60 cm above the floor. Mice were allowed to grasp the wire with their forepaws, thus allowing them to hang from the wire with both forepaws. Each trial was evaluated by a 5-point scale: 0, if the mice fall immediately; 1, if the mice hang via both forelimbs and attempt to climb onto the wire; 3, if mice grasp the wire with both forelimbs and one or both hind limbs while attempting to climb onto to wire; 4, if mice grasp the wire with all four limbs, as well wrapping its tail around the wire; and 5, if the mice escape and reach to either of the poles. Each testing session was 3- successive trials per animal. The average of the three successive trials was reported for each mouse.
Balance Beam walking test
This test was used to assess fine motor coordination following the protocol described earlier by us23. Before surgery, mice were trained to walk on a tapering beam for at least 3 sessions. On the day of testing, the home cage was placed at the end of the narrow end of the beam, and mice were placed on the widest end of the beam. Mice were allowed to cross the beam from the wide end to the narrow end and the time taken to cross the beam was recorded as completion time.
Measurement of relative cerebral blood flow (CBF)
Laser speckle contrast imaging (LSCI) analysis was performed on all mice following the protocol reported earlier24,25,26. Briefly, mice were anesthetized, and an incision was made on the skin overlying the skull. The exposed skulls were scanned using an RFLSI-III LSCI instrument (RWD Life Sciences, San Diego, CA) before SAH, immediately after perforation, and then at 3, 6, 24, and 48 h after SAH. The perfusion values from the acquired images of the ipsilateral hemisphere were normalized with the values from the contralateral hemisphere at all-time points and reported as percent of contralateral relative CBF.
Assessment of complete blood count and RBC deformability
Immediately, before euthanasia at 48 h after SAH, mice were deeply anesthetized with isoflurane, and whole blood was drawn via cardiac puncture. Automatic CBC counting was performed by using the HESKA Element HT5 veterinary Hematology analyzer probe (Heska Corp., Loveland, CO). RBC deformability, reported as the elongation index (EI) was determined by using the RheoScan-AnD300 analyzer (RheoMeditech Inc., Seoul, South Korea). A small volume of 6 µl whole blood was mixed with a highly viscous medium (polyvinylpyrrolidone) and sheared into a cuvette system made of glass. A camera inside the analyzer recorded the diffraction pattern and EI was calculated based on the width and height of each RBC that theoretically ellipse fitted27.
Tissue harvest
After the blood harvest at 48 h after SAH, mice were transcardially perfused with cold PBS (n = 5/group) or perfusion-fixed with PBS followed by 4% PFA (n = 5/group). The brains harvested from perfusion groups were snap-frozen in liquid nitrogen until further processing. The brains harvested from the fixation group were placed in 4% PFA overnight followed by immersion in sucrose. Then the brains were embedded in OCT and snap-frozen until further processing.
Immunoblotting
The ipsilateral side of the harvested brain was homogenized as previously described28. Briefly, brain lysed in ice-cold RIPA buffer (89901 Pierce, Thermo, Rockford, IL) mixed with protease and phosphatase inhibitor (78442 Halt, Thermo, Rockford, IL), and the soluble fraction was isolated by centrifugation at 12,000 rpm for 20 min at 4 °C. Total protein concentration in the cell lysate was determined using BCA kit (23227 Pierce, Thermo, Rockford, IL). Tissue lysate proteins (35 µg) were separated on 4–20% TGX precast linear gradient gels (BioRad labs, Des Plaines, IL) followed by transfer onto nitrocellulose membrane (Biorad laboratories, Germany). Membranes were incubated overnight at 4 °C with the following primary antibodies. AMPK (1:1000 BS 10344R, Bioss, Woburn, MA), AMPK alpha-1/2 (Thr183/Thr172; BS 4002R, Bioss, Woburn, MA), total OXPHOS rodent WB antibody cocktail MS604 (1:1000, ab110413, Abcam Inc, Waltham, MA), OPA1 (1: 1000, MA5-16149, Thermofisher Scientific, Waltham, MA), and mitofusin 2 (Mfn2; 1:1000, 12186-1-AP, Proteintech Group Inc., Rosemont, IL). Followed by three wash membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Then the membranes were washed, and bands were visualized with ECL solution (Clarity western ECL substrate, 1705062 BioRad labs, Des Plaines, IL). Images were captured by using Syngene G: BOX image station (Frederick, MD) equipped with GeneSys, v1.5.6.0. Quantitative analysis of bands was performed with ImageJ software (Version 1.49; NIH). Loading was assessed using β-actin immunoblots.
Cresyl violet staining and assessment of lumen circumference
Harvested brains were cut on cryostat to obtain 25-µm sections. Then the sections were stained with 0.1% (w/v) Cresyl violet following the method we have described earlier29. All sections were imaged and analyzed using Aperio ScanScope CS and Aperio Image Scope software (Leica Biosystems, Cincinnati, OH) respectively. The lumen circumference of MCA in each section was quantified to assess vasospasm20.
Immunofluorescence staining
The immunofluorescence staining was performed as previously described22,29,30. The cryosections were washed for 20 min in 0.1%PBS-TritonX-100, followed by incubation with 1% BSA for 1 h at room temperature. Then the sections were incubated with respective primary antibodies rabbit-anti Iba1 (1:200, Invitrogen, PA5-27436), rabbit-anti-GFAP (1:200, Thermofisher, PA5-16291), mouse anti-Tuj1 (1:100, COVANCE, MMS-435P), rabbit anti-NeuN (1:250, Invitrogen, PA5-78499), rabbit anti-vWF (1:250, Proteintech, 27186-1-AP), and rabbit anti-Tom20 (abcam ab78547) at 4 °C overnight. Thereafter, sections were incubated with Alexa Fluor 568/488 goat anti-mouse/rabbit secondary antibodies (1:500, Thermo Scientific Rockford, IL) followed by mounting on slides using a Vectashield mounting medium with 4′, 6-diamidino-2- phenylindole (DAPI) (H-1200, Vector Laboratories Inc., Newark, CA), and a coverslip. The confocal images were acquired by using Zeiss Axiovert 200 / Axiovert 200 M microscopes. For quantification, five randomly selected fields from the cortex of each animal were analyzed in four nonadjacent sections that were ~ 100 μm apart using ImageJ software.
Detection of trapped erythrocytes in the cerebral arteries
The trapped erythrocytes were detected as previously described31. The brain sections were stained with lectin for 1 h at room temperature followed by incubation with 0.2% sodium borohydride (NaBH4) in PBS for 30 min. After subsequent washings sections were mounted on slides using Vectashield mounting medium with DAPI (H-1200; Vector Laboratories Inc.). The confocal images were acquired and analyzed as described above.
Statistical analysis
Statistical analyses were performed by using GraphPad Prism 9. Neurologic deficit and grip strength scores were analyzed by the non-parametric Kruskal–Wallis test followed by Dunn’s multiple comparison test and are presented as medians with interquartile ranges (25th and 75th percentiles). The remaining data sets were analyzed using a one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. All data are presented as mean ± SEM and P < 0.05 was considered statistically significant.
Results
Post-SAH treatment with PFC improves cerebral blood flow
Mice were anesthetized and CBF was monitored and recorded immediately before and after perforation, then at 3, 6, 24, and 48 h after SAH (Fig. 1A, B). The baseline CBF in SAH and PFC-treated SAH groups was similar; however, there was a significant drop in CBF immediately after the perforation in both groups. At 3 h after the perforation, the CBF tends to improve similarly in both groups. Interestingly, after the injection of PFC at 5 h after SAH, the CBF in the treated group improved substantially as compared with the SAH group.

PFC treatment restricts CBF decline and leukocyte counts after SAH. (A) Representative CBF perfusion images of the SAH and PFC-treated mice immediately after SAH, at 24 h, and 48 h after SAH. A substantial difference in CBF was observed in the SAH vs. SAH + PFC groups. Blue and green circles indicate the region of interest in the ipsilateral and contralateral sides respectively. (B) Analysis of the data reveals a significant decrease in CBF in SAH and PFC-treated groups immediately after the vessel perforation. In both groups, CBF rebounded considerably at 3 h after the perforation. PFC treatment at 5 h (arrow) after the perforation resulted in a rebound of the CBF significantly as compared with the SAH-only group. A significant elevation in (C) neutrophils, (D) monocytes, and (E) basophils counts was observed in the SAH group as compared with the sham group. Interestingly, SAH treatment with PFC decreased the counts of these leukocytes as compared with the SAH group. Data are presented as mean ± SEM, n = 10/group for CBF, n = 6/group for CBC counts. **P < 0.01, ***P < 0.001versus sham group and ##P < 0.01, ###P < 0.001 versus SAH group.
Post-SAH treatment with PFC restricts leukocyte level
The Leukocytes population is reported to increase after injury and trauma. Therefore, to determine leukocyte levels, blood samples were analyzed for CBC using Element HT5 veterinary Hematology Analyzer. We found a significant increase in the counts of some leukocytes such as neutrophils (Fig. 1C), monocytes (Fig. 1D), and basophils (Fig. 1E) in the SAH group. Treatment with PFC significantly decreased the counts of these cells as compared with the SAH group (Fig. 1C-E). No changes were observed in other leukocytes (data not shown).
Post-SAH PFC treatment improves behavioral outcomes and limits cerebral vasospasm
Assessment of the neurological deficits scores revealed significantly higher deficits in the SAH group as compared with the sham group and the treatment with PFC attenuated the neurological deficits as compared to the SAH group (Fig. 2A). Similarly, motor coordination assessment by rotarod (Fig. 2B) and grip strength assessed by hanging wire test (Fig. 2C) also reveal that the SAH group had higher deficits and the treatment with PFC reduced these deficits. Furthermore, fine movement coordination assessed by narrow beam walk also suggests a significant impairment in the SAH group, and the PFC treatment attenuated this impairment significantly (Fig. 2D).

Treatment with PFC improves functional outcomes and prevents vasospasm after SAH. (A) SAH resulted in a significant increase in neurological deficits whereas PFC treatment decreased the neurological deficits. (B) The grip strength test is a measure of dexterity while hanging from a suspended wire. The PFC-treated group showed better scores as compared with the SAH group at 48 h post-SAH. (C) Rotarod testing was done to test motor and coordination activity following SAH in mice. SAH group mice stayed less on the moving rotarod as compared with the sham group. Treatment with PFC improved their retention on the rotarod. (D) The narrow beam-walking test was performed to assess fine motor balance. The SAH group mice took a significantly longer time to cross the narrow beam as compared with the Sham group; whereas the PFC-treated group exhibited a significantly lower time to cross the narrow beam as compared with the SAH group. (E) Representative images of the whole brain exhibiting blood clots after SAH (arrow). The brain from the SAH-only group exhibits a substantial amount of subarachnoid blood 48 h after SAH; whereas the brain from PFC treated group shows only a minimal amount of blood. Representative Cresyl violet staining of the brain sections from the SAH group and PFC-treated group exhibiting (F) changes in middle cerebral artery (MCA) lumen circumference and (G) whole brain sections showing the cite of MCA perforation. (H) Quantitative analysis of the MCA lumen shows a significant decrease in the lumen circumference in SAH brains as compared with the sham brain; whereas the post-SAH treatment with PFC prevented the reduction in lumen circumference as compared with SAH brains. Arrows in panel E show blood clot ___location and the arrow in panel G shows the MCA perforation ___location. Data are expressed as mean ± SEM, n = 10/group for functional assessments and n = 5/group for histological assessments. *P < 0.05, ***P < 0.001 versus sham group. #P < 0.05, ###P < 0.001 versus SAH group. Scale bar = 100 μm.
At 48 h after SAH, harvested brains exhibited blood clots along the circle of the Willis area in SAH as well as PFC-treated SAH groups (Fig. 2E). The blood clot was more profound in the SAH group than in the SAH group treated with PFC. Moreover, Cresyl violet staining of the brain section reveals a significant decrease in the MCA lumen circumference in the SAH group as compared with the sham surgery group, suggesting the onset of vasospasm after SAH (Fig. 2F). Interestingly, the PFC treatment prevented the decrease in lumen circumference, suggesting that PFC treatment after SAH limited the onset of vasospasm (Fig. 2F, H). The endoperforation of the vessel didn’t result in brain infarction or intraparenchymal hematoma (Fig. 2G).
Post-SAH treatment with PFC improves neuronal survival and limits gliosis
We tested class III β-tubulin and NeuN expression to check the neuronal survival after SAH and PFC treatment. The distribution and intensity of Tuj1 were used to assess injury-related alteration in neuronal cytoskeleton. By performing immunofluorescence staining for class III β-tubulin Tuj1 (red) and NeuN (green), we found a significant loss of intensity and co-localization of Tuj1 and NeuN expression in the SAH group as compared to the sham group. Whereas higher intensity and co-localization of Tuj1 with NeuN were found in the post-SAH PFC-treated group as compared with the SAH group (Fig. 3A, B).

PFC treatment after SAH improves neuronal survival and attenuates gliosis. (A) Representative immunofluorescence staining of NeuN (green) and Tuj1(red), GFAP, and Iba1 show changes in neuronal survival, GFAP, and Iba1 immunoreactivity in sham, SAH, and SAH + PFC groups. (B) Quantitative analysis of the NeuN and Tuj1 staining reveals significant neuronal death in the SAH group as compared with the SAH group, whereas treatment with PFC significantly improved neuronal survival as compared with the SAH group. Quantitative analysis of (C) GFAP and (D) Iba1 immunoreactivity exhibits significantly higher gliosis in the SAH group as compared with the sham groups and the post-SAH treatment with PFC reduced gliosis significantly. Data are expressed as mean ± SEM, n = 5/group. **P < 0.01, ***P < 0.001 versus sham group and ##P < 0.01, ###P < 0.001 versus SAH group. Scale bar = 100 μm.
We also tested the potential beneficial effect of PFC treatment after SAH on reactive gliosis. Immunofluorescence staining was performed to test microglial and astrocyte activation in the cerebral cortex at 48 h after SAH. A significant increase in GFAP (Fig. 3A, C) and Iba1 (Fig. 3A, D) expression in the ipsilateral cortex of the SAH group as compared with the sham group. Treatment with PFC significantly reduced the expression of GFAP and Iba1 as compared with the SAH group.
Post-SAH treatment with PFC attenuates vascular impairment and RBC deformability
Following vascular injury, von Willebrand factors (vWFs) bound to subendothelial collagen are exposed. Therefore, to assess the severity of the injury, vWF immunoreactivity and their co-localization with lectin (endothelial marker) were performed. Representative confocal images show the expression of vWF in sham, SAH, and PFC-treated SAH groups (Fig. 4A, B). A significant increase in the expression of vWF was observed in the vasculature of the SAH group as compared with the sham group. Treatment with PFC post-SAH reduced the expression of vWF as compared with the SAH group.

PFC treatment after SAH attenuates vWF expression and RBC entrapment and their deformability. (A) Representative confocal images show different expressions of vWF in sham, SAH, and PFC-treated SAH groups groups. (B) Quantitative analysis of the staining reveals significantly higher expression of vWF in the SAH group as compared with the sham groups, whereas PFC treatment significantly reduced the expression of vWF as compared with the SAH group. (C) Representative fluorescent images of brain sections stained with lectin (green) represent endothelial cells and NaBH4 (red) represents trapped erythrocytes in microvasculature at 48 h after SAH. (D) Quantitative analysis of the sections reveals significantly higher intensities of vascular RBC entrapment in the SAH group as compared with the sham group. Treatment with PFC reduced the number of intensities of entrapped RBC significantly as compared with the SAH group. (E) We also assessed the elongation index to determine RBC deformability at 48 h after SAH. We found a significant reduction in the RBC elongation index in the SAH group. However, treatment with PFC improved the RBC elongation index suggesting that PFC treatment reduced RBC rigidity. Data are expressed as mean ± SEM, n = 5/group. ***P < 0.001 versus sham group, ###P < 0.001 versus SAH group. Scale bar = 100 μm.
Vasospasms in the microvasculature, such as arterioles and capillaries, after SAH may result in the entrapment of erythrocytes. To further ensure that PFC treatment restricted the vasospasm and consequently limited the entrapment of the erythrocytes, brain sections were treated with NaBH4. Sections were stained with lectin to visualize capillaries and NaBH4 to react with trapped erythrocytes resulting in the confirmation of the trapped erythrocytes in the vasculature. There was a significantly higher number of vascular segments exhibiting entrapped erythrocyte intensities in the SAH group as compared to the sham group and the PFC treatment group exhibited lower erythrocyte intensities as compared with the SAH group (Fig. 4C, D).
RBCs are uniquely capable of adapting their shape to the dynamically changing blood flow demands, a condition known as deformability31. Thus, the RBCs’ deformability allows them to transport oxygen under challenging conditions. However, under traumatic conditions, RBCs are reported to lose their deformability which reduces blood flow and tissue oxygenation. RBC deformability is represented in the elongation index (EI). The higher the elongation index, the higher the deformability or lower the rigidity. Under healthy conditions, RBCs (~ 7.0 μm in diameter) flex and elongate to fit the diameter of the capillaries (3.0 to 5.0 μm) under normal endothelial control. Whereas, under disease conditions, a decrease in RBC deformability results in RBC stacking and blockage of the microvessels thus leading to a gradual decrease in CBF and tissue oxygenation leading to tissue ischemia/hypoxia. To test the RBC deformability, whole blood was tested using RheoSCAN. We found a significant decrease in EI in the SAH group as compared with the Sham group, whereas the treatment of the SAH group with PFC significantly increased EI as compared with the SAH group (Fig. 4E).
PFC treatment after SAH inhibits oxidative damage
Previous studies show that oxidative stress augments early brain injury after SAH32. To examine the effect of PFC on oxidative damage, brain sections from the SAH group and PFC-treated SAH groups were used for immunofluorescence staining. To assess oxidative stress, a lipid peroxidation marker 4-HNE, Oxidative DNA damage marker 8-OHdG, and DNA double-strand break marker P-H2A.X were investigated. A significant increase in the oxidative stress markers was observed in the SAH group as compared with the sham group. Treatment with PFC after SAH significantly attenuated the oxidative stress markers as compared with the SAH group (Fig. 5A, B).

PFC treatment attenuates SAH-mediated oxidative stress. (A) Representative immunofluorescence staining images of oxidative damage markers for lipid peroxidation (4-HNE), peroxynitrite production (3-nitrotyrosine, 3-NT), DNA double-strand break (PH2A.X), and oxidized DNA damage (8-OHdG). The fluorescent intensity was quantified using ImageJ analysis software and expressed as percentage changes versus the sham group. (B) Quantitative analysis of the staining reveals a significant increase in oxidative stress markers in the SAH group as compared with the sham group, whereas the treatment with PFC significantly reduced the levels of oxidative stress markers as compared with the SAH group. Data are expressed as mean ± SEM, n = 5/group. *P < 0.05 versus sham group, #P < 0.05 versus SAH group.
PFC treatment preserves mitochondrial morphology and ameliorates their function after SAH
We investigated the effect of post-SAH PFC treatment on mitochondrial morphology following SAH. Quantitative analysis of mitochondrial fragmentation, shown as total particles, revealed a significantly higher degree of SAH-induced mitochondrial fragmentation indicated by the red arrow (Fig. 6A, B). Interestingly, PFC treatment after SAH significantly attenuated mitochondrial fragmentation. For clear image views and quantitative analyses, acquired images of Tom20 fluorescent staining were thresholded, filtered, and binarized, using ImageJ software.

PFC treatment preserves mitochondrial dynamics, mitochondrial oxidative phosphorylation, and fusion protein expression after SAH. To assess mitochondrial fragmentation, brain sections were stained with anti-Tom20 antibody (red) and nuclei were counter-stained with DAPI (blue). Acquired images of Tom20 fluorescent staining were threshold, filtered (median, 2.0 pixel), and binarized for image view, and quantitative analyses were done for each group. (A) Representative confocal microscopy images of immunostained brain sections from the sham, SAH, and PFC-treated SAH groups. Red arrows indicate locations of mitochondrial fragmentation. (B) Quantitative analysis of the staining reveals a significant increase in mitochondrial fragmentation in the SAH group as compared with the sham group, whereas the treatment with PFC significantly reduced the mitochondrial fragmentation as compared with the SAH group. (C) Representative Western blots showing changes in mitochondrial complexes V (ATP5A), IV (MTCO1), II (SDHB), and I (NDUFS8) in sham, SAH, and PFC-treated SAH groups. The representative bands were cropped (box with black lines) from the original bands shown in the Supplementary file, Fig. 1 (B). The exposure times for OXPHOS and β-actin were 15 s. (D) Quantitative analysis of the blots reveals a significant decrease in the investigated mitochondrial complexes in the SAH groups as compared with the sham group, whereas the treatment with PFC significantly improved the expression of these complexes as compared with the SAH group. (E) Representative Western blots showing changes in mitochondrial fusion protein Mfn2 and OPA1 in sham, SAH, and PFC-treated SAH groups. The representative bands were cropped (box with black lines) from the original bands shown in the Supplementary file, Fig. 2 (B). Exposure times for all gels were 15 s. (F) Quantitative analysis of the blots reveals a significant decrease in the investigated mitochondrial fusion proteins in the SAH groups as compared with the sham group, whereas the treatment with PFC significantly improved the expression of these fusion proteins as compared with the SAH group. Red arrows indicate fragmentated parts. Data are expressed as mean ± SEM, n = 5/group. *P < 0.05, **P < 0.01, ***P < 0.001 versus sham group, #P < 0.05, ##P < 0.01, ###P < 0.001 versus SAH group.
We also investigated the effect of PFC treatment on the level of mitochondrial OXPHOS complex V (ATP5A), IV (MTCO1), II (SDHB), I (NDUFS8), and fusion proteins, OPA1 and Mfn2. A significant reduction in the level of OXPHOS complexes at 48 h after SAH was observed, whereas the treatment with PFC improved the expression of these complexes (Fig. 6C, D, and supplementary file). A significant reduction in the expression of Mfn2 fusion proteins was observed in the SAH group as compared to the sham group (Fig. 6E, F, and supplementary file). PFC treatment after SAH upregulated the expression of Mfn2 as compared to the SAH group. There was a decreasing trend in OPA1 expression in the SAH group and an improvement after PFC treatment; however, none of these trends reached a significance level.
Treatment with PFC increased AMPK activation in SAH mice
AMP-activated protein kinase is a key metabolic and stress sensor that is reported to decrease under traumatic conditions. Therefore, we assessed whether PFC treatment improves AMPK phosphorylation after SAH. A significant decrease in phosphorylation of AMPK was observed at 48 h post-SAH as compared with the sham group. Treatment with PFC significantly preserved the phosphorylation of AMPK as compared with the SAH group (Fig. 7A, B, and supplementary file).

PFC treatment restores AMPK phosphorylation after SAH. Phosphorylated AMPKα1 (p-AMPKα1), a measure of AMPK activation, was assessed at 48 h after SAH in mice brain tissue. (A) Representative Western blot images show the changes in pAMPKα1 and total AMPKα1 in sham, SAH, and PFC-treated SAH groups. The representative bands were cropped (box with black lines) from the original bands shown in the Supplementary file, Fig. 3 (A). Exposure times for pAMPK, AMPK, and β-actin were 30, 30, and 15 s respectively. (B) Quantitative analysis of the ratio of pAMPKα1 and total AMPKα1 shows a significant loss of AMPK phosphorylation after SAH. Treatment with PFC improved AMPK phosphorylation. Data are expressed as mean ± SEM, n = 5/group. **P < 0.05 versus sham group, ##P < 0.05 versus SAH group.
Discussion
PFCs are fluorinated biologically inert organic hydrocarbon chains coated with an emulsifier and a stabilizer that can dissolve large amounts of respiratory gases including O233. These properties of PFCs have led to their use as potential RBC alternatives. Moreover, PFC-base emulsions have been used to check their role as oxygen therapeutic in TBI, heart attack, air embolism, and ischemic condition18,19,33,34. A recent study has synthesized a novel PFC oxygen carrier (PFOC) and investigated its role in limiting hypoxic brain injury after SAH35. However, this study didn’t investigate the role of PFC in limiting vascular impairments and vasospasm after SAH. The present study aimed to investigate the feasibility of a specific PFC oxygen carrier, PFC-Oxygent, in limiting brain damage in a perforation model of SAH. The findings from this study indicate that PFC treatment after SAH attenuates functional deficits, limits vasospasm, and attenuates neuronal cell death. The findings also indicate improvement in the expression of the energy sensor AMPK, mitochondrial function, and reduction in oxidative stress following PFC treatment after SAH. As per our knowledge, this is the first study to test the efficacy of PFC-Oxygent in limiting SAH severity.
At the onset of SAH, elevated intracranial pressure leads to an abrupt fall in CBF, acute energy crisis, and neural cell death. After the sudden drop in CBF at the onset of SAH, CBF gradually improves for a short period of time36. However, the onset of vasospasm causes the CBF to decline again. The secondary decline in CBF due to vasospasm eventually leads to DCI which subsequently plays a critical role in the poor outcomes in patients suffering from SAH37,38. Similar patterns were observed in our model where CBF was decreased significantly as compared with the baseline immediately after vascular perforation. The CBF improved at 3 h after SAH but then again started to decline in the SAH group. Similar trends in CBF changes after SAH have been reported36,39. Interestingly, the SAH group treated with PFC exhibited improvement in CBF over time (Fig. 1A, B). Since in animal models, vasospasm is reported to occur around 24 h post-SAH, we observed the secondary decline in CBF at 6 h which reached its lowest at 24 h and remained unchanged thereafter, whereas PFC treatment prevented the onset of vasospasm and limited the secondary fall in CBF beyond 6 h of perforation. Our data also shows the accumulation of blood clots around the area of perforation after SAH. This is further confirmed by the histological data exhibiting a significant decrease in the MCA lumen circumference in the SAH group while the treatment with PFC limits the decrease in MCA lumen circumference (Fig. 2F, H). Based on our data, we believe that PFC treatment prevented the onset of vasospasm.
One of the markers for vascular injury is the expression of vascular von Willebrand Factor (vWF), which is present in Weibel-Palade bodies of endothelial cells and alpha granules of platelets and released in circulation after any activation by injury. It plays an important role in platelet adhesion and aggregation after vascular injury40. Previous studies have documented that vWF mediates leukocyte extravasation and inflammatory response in ischemic cerebral injury41. In the present study, we found that SAH resulted in significantly higher expression of vWF as compared with the sham group, and the SAH group treatment with PFC exhibited significantly lower expression of vWF as compared with the SAH group (Fig. 4A, B). Exposure of the vascular vWF attracts various adhesion cells and leukocytes towards it and forms a vascular plug. Such plugs can also create a barrier in optimal CBF and RBC passage. Therefore, we believe that the aggravated NaBH4 staining observed in this study (Fig. 4C, D) after SAH could be due to enhanced vWF exposure and elevated count of neutrophils and monocytes (Fig. 1) resulting in the development of vascular impairment. Moreover, SAH led to RBC deformability thus causing their entrapment in the impaired capillaries and arterioles (Fig. 4C-E). This limits the oxygen supply to the affected brain regions and leads to neuronal death.
We found a significant increase in neuronal death in the SAH group while PFC treatment after SAH improved neuronal survival (Fig. 3A, B). The massive infiltration of blood in the subarachnoid space and subsequent hemolysis42,43,44,45 generate high levels of hemoglobin (Hb) and other degraded byproducts which augment oxidative stress46,47,48. Oxidative stress has been reported to affect mitochondrial activity and their morphology. In this study, we found that SAH treatment with PFC significantly reduced mitochondrial fragmentation as compared with the SAH group (Fig. 6A, B). Furthermore, neuroinflammation is also a critical component to augment brain damage after SAH. Generally, infiltration of immune cells activates microglia and astrocytes to augment neuroinflammation. We found that PFC treatment after SAH reduced GFAP (Fig. 3A, C) and Iba1 immunoreactivity (Fig. 3A, D), thus potentially limiting inflammation.
ATP is produced in living organisms by a catabolic process of nutrients and the majority of ATP production occurs in mitochondria by oxidative phosphorylation (OXPHOS)14. Normal mitochondrial morphology is vital in the maintenance of mitochondrial function and energy metabolism49. Mitochondrial dysfunction is recognized as a critical pathological factor in augmenting SAH-mediated brain injury32,50. Studies have also shown that SAH can cause mitochondrial dysfunction in the brain17 which can lead to decreased ATP production, increased oxidative stress, and impaired cellular metabolism16,51 We found significant downregulation of OXPHOS at 48 h after SAH. Treatment with PFC improved the expression of OXPHOS, and rescued tissue damage as shown in (Fig. 6C, D).
Under normal conditions, mitochondrial fusion maintains mitochondrial integrity through mitochondrial fusion protein Mfn2 and OPA1. Mitochondrial fusion is reported to have beneficial effects in neurologic conditions. Mfn2 and OPA1 play an indispensable role in regulating mitochondrial fusion and participate in many cellular activities including oxidative stress, cell proliferation, and apoptosis52,53. Mfn254 and OPA155 are reported to decline in preclinical models of SAH and we also observed similar results. We observed a significant decrease in Mfn2 expression after SAH while PFC treatment improved their expression (Fig. 6E, F), while no significant effect was observed in OPA1 level across the groups (Fig. 6E, F). Although fusion proteins play critical roles in various cellular activities, the mechanism(s) that regulate these fusion proteins is still debated.
Based on the role of AMPK as an energy sensor, we tested AMPK phosphorylation after SAH. We found that SAH resulted in a significant loss of AMPK phosphorylation after SAH while PFC treatment preserved the AMPK phosphorylation (Fig. 7A, B). Activation of AMPK is reported to have protective effects on the brain by improving mitochondrial function54. Based on the collective data, we believe that AMPK phosphorylation is partially involved in limiting mitochondrial fusion and oxidative stress after SAH. The mechanism that how PFC increases pAMPK remains to be investigated.
Conclusions
In conclusion, using the endoperforation model we demonstrated that SAH decreases CBF, augments RBCs’ stacking in the microvasculature, and neuronal death. Our data also show that SAH induces neuroinflammation through gliosis, oxidative stress, and mitochondrial dysfunction. Our data also shows that post-SAH treatment with PFC prevents the decline in CBF, attenuates RBCs’ stacking, and reduces neuroinflammation, oxidative stress, and mitochondrial dysfunction. Our data suggests that the beneficial effects of PFC treatment could be partially through the regulation of AMPK phosphorylation, though additional studies are required to establish the beneficial role of PFC-mediated AMPK activation on SAH outcomes.
Limitations of this study
Early brain injury has been implicated in augmenting the severity of DCI and exacerbating the anatomical and functional impairment after SAH. Our data suggests that PFC treatment after SAH minimizes the deleterious cascade of brain injury at early time points which then limits the onset of vasospasm and potentially DCI. However, additional studies are required to establish the role of PFC treatment in limiting vasospasm and DCI after SAH by performing long-term studies. This study also tested the effect of a single-dose treatment given at a single time point. A dose-response curve and treatment window studies are required to fully unravel the therapeutic potential of PFC. This study was also limited by testing the efficacy of PFC in limiting the severity of SAH in male mice only. Further studies are required to test the efficacy of PFC in female mice. Nevertheless, this study reports for the first time the beneficial effects of an oxygen carrier in limiting the severity of SAH.
Data availability
All data generated and analyzed during this study are available from the corresponding author upon reasonable request.
References
Lantigua, H. et al. Subarachnoid hemorrhage: who dies, and why? Crit. Care 19, 309. https://doi.org/10.1186/s13054-015-1036-0 (2015).
Fujii, M. et al. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl. Stroke Res. 4, 432–446. https://doi.org/10.1007/s12975-013-0257-2 (2013).
Macdonald, R. L. et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 10, 618–625. https://doi.org/10.1016/S1474-4422(11)70108-9 (2011).
Macdonald, R. L. et al. Randomized trial of clazosentan in patients with aneurysmal subarachnoid hemorrhage undergoing endovascular coiling. Stroke 43, 1463–1469. https://doi.org/10.1161/STROKEAHA.111.648980 (2012).
Macdonald, R. L., Pluta, R. M. & Zhang, J. H. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat. Clin. Pract. Neurol. 3, 256–263. https://doi.org/10.1038/ncpneuro0490 (2007).
Kassell, N. F., Sasaki, T., Colohan, A. R. & Nazar, G. Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke 16, 562–572. https://doi.org/10.1161/01.str.16.4.562 (1985).
Kahn, B. B., Alquier, T., Carling, D. & Hardie, D. G. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell. Metab. 1, 15–25. https://doi.org/10.1016/j.cmet.2004.12.003 (2005).
Hill, J. L. et al. Traumatic brain injury decreases AMP-activated protein kinase activity and pharmacological enhancement of its activity improves cognitive outcome. J. Neurochem. 139, 106–119. https://doi.org/10.1111/jnc.13726 (2016).
Chen, S. et al. Activation of melanocortin receptor 4 with RO27-3225 attenuates neuroinflammation through AMPK/JNK/p38 MAPK pathway after intracerebral hemorrhage in mice. J. Neuroinflam. 15, 106. https://doi.org/10.1186/s12974-018-1140-6 (2018).
Huang, Y. et al. Puerarin attenuates oxidative stress and ferroptosis via AMPK/PGC1alpha/Nrf2 pathway after subarachnoid hemorrhage in rats. Antioxid. (Basel) 11 https://doi.org/10.3390/antiox11071259 (2022).
Macdonald, R. L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 10, 44–58. https://doi.org/10.1038/nrneurol.2013.246 (2014).
Forrester, S. J., Kikuchi, D. S., Hernandes, M. S., Xu, Q. & Griendling, K. K. Reactive oxygen species in metabolic and inflammatory signaling. Circ. Res. 122, 877–902. https://doi.org/10.1161/CIRCRESAHA.117.311401 (2018).
Hinchy, E. C. et al. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 293, 17208–17217. https://doi.org/10.1074/jbc.RA118.002579 (2018).
Herzig, S. & Shaw, R. J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell. Biol. 19, 121–135. https://doi.org/10.1038/nrm.2017.95 (2018).
Spiess, B. D. Perfluorocarbon gas transport: an overview of medical history with yet unrealized potentials. Shock 52, 7–12. https://doi.org/10.1097/SHK.0000000000001150 (2019).
Lin, M. T. & Beal, M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. https://doi.org/10.1038/nature05292 (2006).
Jacobsen, A., Nielsen, T. H., Nilsson, O., Schalen, W. & Nordstrom, C. H. Bedside diagnosis of mitochondrial dysfunction in aneurysmal subarachnoid hemorrhage. Acta Neurol. Scand. 130, 156–163. https://doi.org/10.1111/ane.12258 (2014).
Daugherty, W. P., Levasseur, J. E., Sun, D., Spiess, B. D. & Bullock, M. R. Perfluorocarbon emulsion improves cerebral oxygenation and mitochondrial function after fluid percussion brain injury in rats. Neurosurgery 54, 1223–1230. https://doi.org/10.1227/01.neu.0000119238.68938.5d (2004). discussion 1230.
Torres, L. N., Spiess, B. D. & Torres Filho, I. P. Effects of perfluorocarbon emulsions on microvascular blood flow and oxygen transport in a model of severe arterial gas embolism. J. Surg. Res. 187, 324–333. https://doi.org/10.1016/j.jss.2013.08.011 (2014).
Kamat, P. K., Ahmad, A. S. & Doré, S. Carbon monoxide attenuates vasospasm and improves neurobehavioral function after subarachnoid hemorrhage. Arch. Biochem. Biophys. 676, 108117. https://doi.org/10.1016/j.abb.2019.108117 (2019).
Clark, W., Gunion-Rinker, L., Lessov, N. & Hazel, K. Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke 29, 2136–2140 (1998).
Ahmed, M. E. et al. Glia maturation factor (GMF) regulates microglial expression phenotypes and the associated neurological deficits in a mouse model of traumatic brain Injury. Mol. Neurobiol. 57, 4438–4450. https://doi.org/10.1007/s12035-020-02040-y (2020).
Ahmed, M. E. et al. Methylene blue promotes cortical neurogenesis and ameliorates behavioral deficit after photothrombotic stroke in rats. Neuroscience 336, 39–48. https://doi.org/10.1016/j.neuroscience.2016.08.036 (2016).
Lima, B. et al. Endogenous S-nitrosothiols protect against myocardial injury. Proc. Natl. Acad. Sci. U. S. A. 106, 6297–6302. https://doi.org/10.1073/pnas.0901043106 (2009).
Hoda, M. N. et al. Remote ischemic perconditioning is effective after embolic stroke in ovariectomized female mice. Transl. Stroke Res. 5, 484–490. https://doi.org/10.1007/s12975-013-0318-6 (2014).
Zaidi, S. K., Ahmed, F., Alkhatabi, H., Hoda, M. N. & Al-Qahtani, M. Nebulization of low-dose S-Nitrosoglutathione in diabetic stroke enhances benefits of reperfusion and prevents post-thrombolysis Hemorrhage. Biomolecules 11 https://doi.org/10.3390/biom11111587 (2021).
Moon, J. S. et al. Impaired RBC deformability is associated with diabetic retinopathy in patients with type 2 diabetes. Diabetes Metab. 42, 448–452. https://doi.org/10.1016/j.diabet.2016.04.008 (2016).
He, T. et al. Subarachnoid hemorrhage enhances the expression of TDP-43 in the brain of experimental rats and human subjects. Exp. Ther. Med. 16, 3363–3368. https://doi.org/10.3892/etm.2018.6636 (2018).
Ahmad, A. S., Mendes, M., Hernandez, D. & Dore, S. Efficacy of Laropiprant in minimizing brain Injury following experimental intracerebral hemorrhage. Sci. Rep. 7, 9489. https://doi.org/10.1038/s41598-017-09994-5 (2017).
Lu, Y. et al. Low-level laser therapy for beta amyloid toxicity in rat hippocampus. Neurobiol. Aging 49, 165–182. https://doi.org/10.1016/j.neurobiolaging.2016.10.003 (2017).
Yemisci, M. et al. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 15, 1031–1037. https://doi.org/10.1038/nm.2022 (2009).
Zhang, T. et al. Docosahexaenoic acid alleviates oxidative stress-based apoptosis Via improving mitochondrial dynamics in early Brain injury after subarachnoid hemorrhage. Cell. Mol. Neurobiol. 38, 1413–1423. https://doi.org/10.1007/s10571-018-0608-3 (2018).
Riess, J. G. Fluorocarbon-based in vivo oxygen transport and delivery systems. Vox Sang. 61, 225–239. https://doi.org/10.1111/j.1423-0410.1991.tb00952.x (1991).
Stowell, C. P., Levin, J., Spiess, B. D. & Winslow, R. M. Progress in the development of RBC substitutes. Transfusion 41, 287–299. https://doi.org/10.1046/j.1537-2995.2001.41020287.x (2001).
Peng, Z. et al. Novel perfluorocarbon-based oxygenation therapy alleviates Post-SAH hypoxic brain injury by inhibiting HIF-1α. Free Radic. Biol. Med. 214, 173–183. https://doi.org/10.1016/j.freeradbiomed.2024.02.002 (2024).
Bederson, J. B., Germano, I. M. & Guarino, L. Cortical blood flow and cerebral perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke 26, 1086–1092. https://doi.org/10.1161/01.STR.26.6.1086 (1995).
Cahill, J., Calvert, J. W. & Zhang, J. H. Mechanisms of early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 26, 1341–1353. https://doi.org/10.1038/sj.jcbfm.9600283 (2006).
Ostrowski, R. P., Colohan, A. R. & Zhang, J. H. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol. Res. 28, 399–414. https://doi.org/10.1179/016164106X115008 (2006).
Sehba, F. A. Rat endovascular perforation model. Transl. Stroke Res. 5, 660–668. https://doi.org/10.1007/s12975-014-0368-4 (2014).
Fan, X., Lo, E. H. & Wang, X. Effects of minocycline plus tissue plasminogen activator combination therapy after focal embolic stroke in type 1 diabetic rats. Stroke 44, 745–752, https://doi.org/10.1161/STROKEAHA.111.000309
Chauhan, A. K. et al. ADAMTS13: a new link between thrombosis and inflammation. J. Exp. Med. 205, 2065–2074. https://doi.org/10.1084/jem.20080130 (2008).
Kusaka, G., Ishikawa, M., Nanda, A., Granger, D. N. & Zhang, J. H. Signaling pathways for early brain injury after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 24, 916–925. https://doi.org/10.1097/01.WCB.0000125886.48838.7E (2004).
Suarez, J. I. Timing of neuropsychological outcome measures in patients with subarachnoid hemorrhage. Stroke 38, 1724–1725. https://doi.org/10.1161/STROKEAHA.107.487181 (2007).
Connolly, E. S. Jr. et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American heart association/american stroke Association. Stroke 43, 1711–1737. https://doi.org/10.1161/STR.0b013e3182587839 (2012).
Sun, B., Yang, S., Li, S. & Hang, C. Melatonin upregulates nuclear factor erythroid-2 related factor 2 (Nrf2) and mediates mitophagy to protect against early brain Injury after subarachnoid hemorrhage. Med. Sci. Monit. 24, 6422–6430. https://doi.org/10.12659/MSM.909221 (2018).
Dorsch, N. W. & King, M. T. A review of cerebral vasospasm in aneurysmal subarachnoid haemorrhage part I: incidence and effects. J. Clin. Neurosci. 1, 19–26. https://doi.org/10.1016/0967-5868(94)90005-1 (1994).
Mori, T. et al. Intracisternal increase of superoxide anion production in a canine subarachnoid hemorrhage model. Stroke 32, 636–642. https://doi.org/10.1161/01.str.32.3.636 (2001).
Vergouwen, M. D., de Haan, R. J., Vermeulen, M. & Roos, Y. B. Effect of statin treatment on vasospasm, delayed cerebral ischemia, and functional outcome in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis update. Stroke 41, e47–52, https://doi.org/10.1161/STROKEAHA.109.556332
Detmer, S. A. & Chan, D. C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell. Biol. 8, 870–879. https://doi.org/10.1038/nrm2275 (2007).
Huang, L. et al. Inhibitory effects of p38 inhibitor against mitochondrial dysfunction in the early brain injury after subarachnoid hemorrhage in mice. Brain Res. 1517, 133–140. https://doi.org/10.1016/j.brainres.2013.04.010 (2013).
Balog, J., Mehta, S. L. & Vemuganti, R. Mitochondrial fission and fusion in secondary brain damage after CNS insults. J. Cereb. Blood Flow. Metab. 36, 2022–2033. https://doi.org/10.1177/0271678X16671528 (2016).
Zuchner, S. et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-tooth neuropathy type 2A. Nat. Genet. 36, 449–451. https://doi.org/10.1038/ng1341 (2004).
Nasrallah, C. M. & Horvath, T. L. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 10, 650–658. https://doi.org/10.1038/nrendo.2014.160 (2014).
Wu, X. et al. SIRT3 protects against early brain injury following subarachnoid hemorrhage via promoting mitochondrial fusion in an AMPK dependent manner. Chin. Neurosurg. J. 6, 1. https://doi.org/10.1186/s41016-019-0182-7 (2020).
Zhang, Y. et al. Chemerin reverses neurological impairments and ameliorates neuronal apoptosis through ChemR23/CAMKK2/AMPK pathway in neonatal hypoxic-ischemic encephalopathy. Cell. Death Dis. 10, 97. https://doi.org/10.1038/s41419-019-1374-y (2019).
Acknowledgements
This work was supported by NIH grant R21NS123531 (ASA) and the Department of Neurology at Henry Ford Health. The authors are grateful to Professor Bruce Spiess, Vice Chair of Research, Department of Anesthesiology, University of Florida, for his generous gift of PFC-Oxygent.
Author information
Authors and Affiliations
Contributions
M.E.A: methodology, formal analysis, investigation, data curation, writing original draft, visualization. N.A: methodology, writing original draft, visualization. S. F.: methodology, review, and editing. S. A.: conceptualization, writing review, and editing. S. G.: conceptualization, writing review and editing, resources. M. N. H.: conceptualization, methodology, resources. A. S. A.: conceptualization, methodology, investigation, data curation, visualization, investigation, writing review and editing, resources, project administration, supervision. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ahmed, M.E., Akhter, N., Fatima, S. et al. Therapeutic utility of Perfluorocarbon Oxygent in limiting the severity of subarachnoid hemorrhage in mice. Sci Rep 14, 26638 (2024). https://doi.org/10.1038/s41598-024-77321-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-77321-w
