
Abstract
Despite improvements in treatment in recent years, patients with lung adenocarcinoma (LUAD) still face poor prognoses. In this study, we elucidated the potential role of NOP58 ribonucleoprotein in pan-cancer and validated its oncogenic significance in LUAD using bioinformatics and in vitro and in vivo functional assays. NOP58 was found to be overexpressed in various types of tumors. It had great precision for predicting 20 distinct cancer types using receiver operating characteristic curve (ROC) as well as significant connections with the prognoses in particular cancers. In LUAD, NOP58 expression was correlated substantially with the TNM stage, pathologic stage, smoking status, and effectiveness endpoints, when we analyzed its association with clinical characteristics in LUAD. Elevated NOP58 expression was shown as connected with Th2 cell infiltration while also negatively linked with infiltrating other immune cells, such as CD8 T, cytotoxic, and Th1. By inhibiting NOP58 within the LUAD cells, we found a decrease in cells’ capability to proliferate, migrate, and invade. Knockdown of NOP58 inhibited tumor growth in mouse xenograft models. Furthermore, the tissue microarray study indicated that there was a greater expression of NOP58 in the tumor tissues compared to adjacent normal tissues in LUAD. Our findings revealed that NOP58 could be an outstanding bio-index for pan-cancer diagnosis and prognosis and an independent prognostic risk factor in LUAD.
Similar content being viewed by others


Introduction
The incidence and mortality rate of lung cancer are the highest of all cancers worldwide. With 2.09 million deaths annually, lung cancer results continue to remain dismal1. Lung cancer might be separated into small- and non-small cell lung cancer (NSCLC)2. Lung adenocarcinoma (LUAD) is the most prevalent subtype of NSCLC3, with 50% of all lung cancer diagnoses, and its prevalence has been increasing2,4. New treatments for LUAD, including targeted therapy and immunotherapy, have been developed recently4. However, very few individuals suffering from LUAD benefit from these therapies. Cancers continue to change and acquire resistance mechanisms to escape treatment. The need for more effective biomarkers to facilitate novel therapeutics is therefore essential.
The NOP58 ribonucleoprotein is a protein-coding gene located on chromosome 2 that is also known as NOP5, HSPC120, and NOP5/NOP585. This gene encodes a protein of 59.6 kDa molecular weight that is mostly localized in the nucleolus5. NOP58 produces tiny nucleolar ribonucleoprotein complexes with short nucleolar RNAs and facilitates post-transcriptional alterations, pseudouridylation, methylation, and cleavage for rRNA maturation as a nucleolar protein6. NOP58, NOP56, and 15.5 kDa are necessary for carcinogenesis, snoRNA development, stability, and localization7. Overexpression of NOP58 is inversely linked with overall survival (OS) in hepatocellular carcinoma (HCC) individuals and might be a viable bio-index for predicting HCC prognosis8. And it might also contribute to LUAD recurrence9. A study has shown that long noncoding RNA (lncRNA) ZFAS1 could promote small nucleolar RNA-stimulated 2’-O-methylation through recruiting NOP58 in colorectal cancer10. PGM5P3-AS1 reduced the development of triple negative breast cancer (TNBC) by enlisting NOP58 to stabilize the mRNA for MAP1LC3C11. Additionally, another study has shown that NOP58 might enhance the stability of FAM83A mRNA and contribute to the advancement of hepatocellular carcinoma12.Despite NOP58’s molecular actions being observed in these studies, its role in LUAD is not well understood. In this investigation, using bioinformatics, we elucidated the genetic expression, prognostic and diagnostic value, protein phosphorylation, genetic alteration, immune cell infiltration and other characteristics of NOP58. A clinical tissue microarray (TMA), and functional tests using NOP58-knockdown LUAD cells, were used to investigate the clinical importance and possible roles of NOP58 in LUAD. In addition, our study investigated the probable carcinogenic mechanism of the NOP58 gene and its prognostic significance for LUAD.
Results
NOP58 Expression patterns in pan-cancer
Oncomine mRNA levels were elucidated to investigate NOP58 expression in distinct malignancies. Brain and central nervous system (CNS), breast, esophageal, gastric, liver, colorectal, head and neck, and ovarian cancers, as well as leukemia and lymphoma, showed greater NOP58 expression than normal tissue. Only the breast cancer dataset demonstrated a lower NOP58 expression level (Figure S1A). The expression of NOP58 in different malignancies was presented in Table S1.
To further study NOP58 expression in pan-cancer, we assessed Genotype-Tissue Expression (GTEx) and The Cancer Genome Atlas (TCGA) RNA sequencing data using R tools. We compared the patterns of NOP58 expression in cancerous and matching normal tissues in Figure S1B. In breast invasive carcinomas (BRCA), cholangiocarcinoma (CHOL), colon adenocarcinoma (COAD), lymphoid neoplasm diffuse large B-cell lymphoma (DLBC), esophageal carcinoma (ESCA), glioblastoma multiforme (GBM), head and neck squamous carcinoma (HNSC), kidney renal clear cell carcinoma (KIRC), brain lower grade glioma (LGG), liver hepatocellular carcinoma (LIHC), lung squamous cell carcinoma (LUSC), pancreatic adenocarcinoma (PAAD), rectum adenocarcinoma (READ), stomach adenocarcinoma (STAD), testicular germ cell tumors (TGCT), thymoma (THYM), uterine corpus endometrial carcinoma (UCEC), and uterine carcinosarcoma (UCS), NOP58 expression was significantly higher. In contrast, NOP58 expression was considerably lower in kidney chromophobe (KICH), ovarian serous cystadenocarcinoma (OV), thyroid carcinoma (THCA) than in normal tissues (all P less than 0.001). Analyses of the differential NOP58 expression in tumor and non-tumorous tissues produced comparable findings (Figure S1C). The NOP58 levels were elevated in several tumor types including CHOL, ESCA, BRCA, HNSC, COAD, KIRC, LUAD, LIHC, LUSC, prostate adenocarcinoma (PRAD), READ, UCEC and STAD.
Clinical proteomic tumor analysis consortium (CPTAC) dataset demonstrated that breast cancer, ovarian cancer, colon cancer, clear cell renal cell carcinoma (clear cell RCC), LUAD (Figure S1D, P < 0.0001) and UCEC (P < 0.001) had greater NOP58 total protein expression than normal tissues. Additionally, we elucidated the association of NOP58 expression with cancer pathology stages using the “pathological stage plot” module of the Gene Expression Profiling Interactive Analysis 2 (GEPIA2). Adrenocortical carcinoma (ACC), LIHC, KICH, LUAD, STAD, LUSC, THCA, and OV tissues expressed NOP58 mRNA significantly, whereas other types of cancers did not (Figure S1E, P < 0.05).
Date of protein phosphorylation analysis
In addition, the NOP58 phosphorylation levels were examined between primary tumor and normal tissues. The data from CPTAC was utilized to investigate the NOP58 phosphorylation locations and significant changes between six kinds of malignancies (breast cancer, colon cancer, clear cell RCC, LUAD, and ovarian cancer). All primary tumor tissues had a greater level of phosphorylation at the S502 ___location compared to normal tissues (Figure S2, all P < 0.05), except for ovarian cancer (Figure S3, P > 0.05). A high S514 site phosphorylation level for breast cancer, colon cancer and uterine corpus endometrial carcinoma (P < 0.001) was shown in Figure S2. Further, the phosphorylation levels of S502T508, S502S509, S502T510, S502S514, T508, T508S514, T508T510, T508S509, S509T510, S509S514 and T510S514 sites were greater in some tumor tissues than in normal tissues.
Genetic alteration analysis of NOP58 in pan-cancer
We then curated a genetic alteration analysis of NOP58 in malignancies using the cBioPortal database. Mutations were the primary DNA alteration in the TCGA pan-cancer panel. Mutations were found primarily on UCEC, COAD, BLCA, UCS, and STAD (Figure S4A). Another pan-cancer panel revealed that the most frequent DNA alteration was amplification. Amplifications of NOP58 were observed in melanoma, PAAD, LUSC, and PRAD patients (Figure S4B). Furthermore, the sites, case numbers and types of the NOP58 gene modification were presented (Table S2). The NOP58 frameshift deletion mutation was the primary alteration type, while the most frequent mutation was K524Rfs*22 (Figure S5A and S5B). Gain function, diploidy, and shallow deletion were the most common putative copy-number alterations of NOP58 (Figure S5C). In the altered group, TRAJ6, BCDIN3D-AS1, AQP5-AS1, LINC02395, LINC02396, SNORA2A, ADCY6-DT, SNORA2C, and GUCY2GP were more commonly altered than in the unaltered group (Figure S5D).
Functional enrichment for NOP58-associated genes
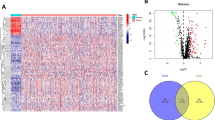
The NOP58-binding proteins and expression-correlated genes were screened for pathway enrichment analyses to examine the molecular mechanism of NOP58 gene expression in carcinogenesis. Using the STRING program, we discovered 40 NOP58-binding proteins validated by experimental data. As Fig. 1A depicts, the TCGA tumor expression data was combined with the GEPIA2 algorithm to identify the top 100 genes that correlate with NOP58 levels. As demonstrated in Fig. 1B, NOP58 levels were positively linked with the WDR12 (R = 0.73), WDR75 (R = 0.72), SRSF7 (R = 0.7), DNAJC2 (R = 0.66), and ORC2 (R = 0.66) genes (all P < 0.0001). NOP58 and the five genes indicated above were positively associated with the most detailed cancer types, according to heatmap data (Fig. 1C). The intersection assessment of the two cohorts indicated that they share seven members, namely, NOP56, MPHOSPH10, DKC1, NOLC1, WDR75, HEATR1 and WDR43 (Fig. 1D). Combining the two datasets allowed us to analyze Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment (Table S3). According to the KEGG data in Fig. 1E, “ribosome biogenesis in eukaryotes,” “spliceosome,” “cell cycle,” and “DNA replication” might be implicated in the influence of NOP58 on the pathogenesis of tumors. The majority of these genes were involved in RNA catalytic, snoRNA binding, DNA-dependent ATPase helicase, and DNA replication activities origin binding, according to the findings of the GO enrichment analysis (Fig. 1E).

NOP58-related gene enrichment analysis. (A) Using STRING, we first obtained the experimentally determined NOP58-binding proteins. (B) We also obtained the top 100 NOP58-correlated genes in TCGA projects. The expression correlation between NOP58 and selected targeting genes, including WDR12, WDR75, DNAJC2, ORC2 and SRSF7 was analyzed using the GEPIA2. (C) Heatmaps were displayed for the detailed cancer types. (D) Intersection analysis of NOP58-binding and correlated genes was performed. (E) KEGG pathway analysis and GO analysis were conducted based on NOP58-binding and interacted genes. TCGA, The Cancer Genome Atlas; GEPIA2, Gene Expression Profiling Interactive Analysis 2; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology.
Survival prognosis analysis
As seen in Fig. 2A-I, increased NOP58 levels were linked with a worse OS prognosis for malignancies including breast cancer (P = 0.021), esophageal adenocarcinoma (P < 0.0001), HNSC (P = 0.044), KIRC (P < 0.0001), kidney renal papillary cell carcinoma (KIRP) (P < 0.0001), LIHC (P < 0.0001), LUAD (P = 0.027), pancreatic ductal adenocarcinoma (P = 0.0027), and UCEC (P = 0.0023) of TCGA datasets. Relapse-free survival (RFS) analysis revealed that elevated NOP58 levels were linked with a worse prognosis in TCGA instances of breast cancer (P = 0.026), KIRP (P = 0.00069), LIHC (P = 0.00013), pancreatic ductal adenocarcinoma (P = 0.028), and UCEC (P = 0.0052) (Fig. 2J-P). The findings provided above demonstrated the association of NOP58 levels with the individuals’ prognosis with various malignancies.

Survival prognosis analysis of cancers. (A-I) OS analysis of NOP58 gene in the TCGA dataset. (J-P) RFS analysis of NOP58 gene in the TCGA dataset. OS, overall survival; RFS, relapse-free survival; TCGA, The Cancer Genome Atlas.
Diagnostic value of NOP58 in pan-cancer
The diagnostic utility of NOP58 in pan-cancer (TCGA) was evaluated using a receiver operating characteristic curve (ROC). NOP58 was shown to be capable of predicting 20 distinct cancer types, including CHOL (area under the curve (AUC) = 1), COAD (AUC = 0.945), COADREAD (AUC = 0.938), READ (AUC = 0.916), ESCA (AUC = 0.932), HNSC (AUC = 0.833), KICH (AUC = 0.930), LIHC (AUC = 0.904), LUAD (AUC = 0.895), LUSC (AUC = 0.952), oral squamous cell carcinoma (OSCC) (AUC = 0.819), esophageal carcinoma (ESAD) (AUC = 0.939), STAD (AUC = 0.976), BRCA (AUC = 0.743), KIRC (AUC = 0.714), PRAD (AUC = 0.764), THCA (AUC = 0.781), bladder cancer (BLCA) (AUC = 0.675) and UCEC (AUC = 0.680), with a certain level of accuracy (AUC > 0.7) (Figures S6A-T). As for CHOL, COAD, COADREAD, READ, ESCA, KICH, LIHC, LUSC, ESAD, and STAD, NOP58 had the highest accuracy (AUC > 0.9).
NOP58 Expression and therapeutic response association among various types of cancer
Chemotherapy responses in clinical cancer cohorts were evaluated concerning NOP58 expression. Breast cancer patients with greater NOP58 levels were more chemotherapy resistant, while those with lower NOP58 levels were more responsive to chemotherapy (Figure S7A). Additionally, reduced NOP58 expression in LGG and UCEC was associated with the therapeutic advantages of immune checkpoint inhibitors (PD-1 or PD-L1). Hence, compared with the cohort with a high expression level of NOP58, the survival period of the low-level cohort was longer (Figure S7B). Furthermore, the level of cytotoxic T lymphocytes was negatively associated with increased NOP58 expression in LGG cancer cohorts, suggesting potential interactions of NOP58 expression with T cell exclusion (P = 0.00668).
NOP58 correlates with different clinical characteristics in LUAD
The NOP58 levels were markedly associated with TNM and pathologic stages of LUAD when we analyzed its association with various clinical characteristics in LUAD. NOP58 was expressed higher in patients with T4 and pathologic stage II/III/IV (Table S4). Furthermore, we elucidated the expression difference of NOP58 in various clinical groups of LUAD. The data showed that NOP58 levels were higher in the subgroup of the T4 stage, N2/N3 stage, M1 stage, and the subgroup of pathologic stage III (Fig. 3A-D). Compared with the NOP58 expression in the subgroup of patients who get complete remission (CR) from the primary therapy, it was higher in the subgroup of patients who get partial remission (PD) (Fig. 3E). Additionally, the expression was also elevated in the subgroup of smokers (Fig. 3F). When we focused on the OS event, and the disease-specific survival (DSS) event, the NOP58 expression in the dead and alive subgroups was different and higher in the dead subgroups (Fig. 3G and 3 H).

Associations between NOP58 expression and different clinical characteristics in LUAD. (A) T stage; (B) N stage; (C) M stage; (D) pathologic stage; (E) primary therapy outcome; (F) smoker; (G) OS event; (H) DSS event (* P < 0.05; ** P < 0.01; *** P < 0.001). LUAD, lung adenocarcinoma; OS, overall survival; DSS, disease-specific survival.
LUAD cox regression analyses (Univariate and Multivariate)
Using univariate and multivariate Cox regression, NOP58 and clinical features of LUAD were studied which revealed that primary treatment results and residual tumor were strongly linked with OS (Table S5), while primary therapy was substantially related to DSS (Table S6). In addition, progression free interval (PFI) was substantially related to the T stage, initial therapeutic outcome, and tumor burden (Table S7).
NOP58 expression and immune characteristics correlation
We evaluated immune infiltration in LUAD with various NOP58 levels using the tumor immune estimating resource (TIMER) database. B cells, dendritic cells (DCs), cytotoxic cells, immature dendritic cells (iDC), eosinophils, macrophages, NK CD56bright cells, natural killer (NK) cells, plasmacytoid dendritic cells (pDC), neutrophils, CD8 T cells, T cells, effector memory T cell (Tem), mast cells, follicular helper T cell (TFH) and Th1 cells in LUAD patients with elevated NOP58 levels were found much lower than those with low NOP58 levels. The infiltration of T helper cells, γδT cells, and Th2 cells was larger in LUAD patients with high NOP58 expression than in those with low NOP58 expression. The levels of infiltration of activated DCs, NK CD56dim cells, Tcm cells, Th17 cells, and Treg cells did not change significantly between individuals with elevated and reduced NOP58 levels (Figure S8).
Furthermore, we elucidated the link between NOP58 levels and immunological infiltration in LUAD and indicated that NOP58 levels were positively connected with Th2 cell infiltration (r = 0.35, P < 0.001) (Figure S8G). In contrast, NOP58 expression was adversely associated with cytotoxic cells (r = -0.150, P < 0.001) (Figure S8C), CD8 T cells (r = -0.141, P = 0.001) (Figure S8D), NK cells (r = -0.109, P = 0.012) (Figure S8E), pDC cells (r = -0.212, P < 0.001) (Figure S8F), Th1 cells (r = -0.166, P < 0.001) (Figure S8H), TFH cells (r = -0.216, P < 0.001) (Figure S8I) and Tem cells (r = -0.109, P = 0.011) (Figure S8J) infiltration. These results indicated that NOP58 might inhibit the immune response to LUAD tumors by favorably regulating Th2 infiltration and negatively regulating cytotoxic cells, CD8 + T-cells, pDC cells, TFH cells, and Tem cells.
The association between NOP58 levels and immune cell chemokines and chemokine receptors in LUAD was investigated using TISIDB. LUAD expression of NOP58 was substantially linked with several chemokines and their receptors, as seen by the heatmap (Fig. 4A and 4B). Expression of NOP58 was shown to be adversely linked with CCL14 (r = − 0.312, P < 4.98e − 13), CX3CR1 (r = − 0.47, P < 2.2e − 16), CCR1 (r = − 0.3, P < 3.88e − 12), CCR2 (r = − 0.402, P < 2.2e − 16), CCR6 (r = − 0.398, P < 2.2e − 16) and CCR7 (r = − 0.302, P < 3.17e − 12) (Fig. 4C–H), while NOP58 was not strongly linked with other chemokines/chemokine receptors (-0.3 < r < 0.3). The expression of NOP58 in LUAD was inversely proportional to the expression of chemokine and chemokine receptors.

Correlation analysis between NOP58 expression and chemokines and/or chemokine receptors. (A) Heatmap analysis revealed the correlation between NOP58 and chemokines in tumors. (B) Heatmap analysis revealed the correlation between NOP58 and chemokine receptors in tumors. (C–H) NOP58 expression LUAD was negatively correlated with CX3CL1, CX3CR1, CCR1, CCR2, CCR6 and CCR7. LUAD, lung adenocarcinoma.
Immune checkpoint inhibitors have been used for tumor immunotherapy for a variety of malignancies with steady improvement in prognosis13. With the help of TISIDB database, the association between NOP58 and the expression of immunoinhibitors and immunostimulators in diverse forms of human cancer were explored (Fig. 5A and 5B). Several immunoinhibitors and immunostimulators were adversely correlated with the expression of NOP58, such as CSF1R (r = − 0.312, P < 4.98e − 13), HAVCR2 (r = − 0.47, P < 2.2e − 16), TGFB1 (r = − 0.3, P < 3.88e − 12), TMEM173 (r = − 0.402, P < 2.2e − 16), C10orf54 (r = − 0.398, P < 2.2e − 16) and CD28 (r = − 0.302, P < 3.17e − 12) in LUAD (Fig. 5C–H). Consequently, NOP58 might regulate tumor immunity, based on these results.

Correlation analysis between NOP58 expression and immunoinhibitors and immunostimulators. (A) Heatmap analysis revealed a correlation between NOP58 and immunoinhibitors in tumors. (B) Analysis of heatmaps to determine a correlation between NOP58 and immunostimulators in tumors. (C–E) NOP58 expression was negatively correlated with the immunoinhibitors CSF1R, HAVCR2 and TGFB1 in LUAD. (F–H) NOP58 expression was negatively correlated with the immunostimulators CD28, TMEM173 and C10orf54 in LUAD. LUAD, lung adenocarcinoma.
Expression of NOP58 in LUAD tissues
To analyze the clinical importance of NOP58 in LUAD, we used TMA to identify the expression of NOP58 in LUAD samples (n = 80) and matched neighboring normal tissues (n = 80) in order to evaluate the clinical significance of NOP58. It showed that LUAD samples exhibited higher NOP58 expression than adjacent normal tissues (Fig. 6A). According to the Human Protein Atlas (HPA) dataset, NOP58 protein expression was likewise elevated in LUAD tissues (Fig. 6B).

The protein expression of NOP58 in immunohistochemical images of HPA database and tissue microarray. (A) NOP58 protein expression levels in LUAD and normal lung tissues (HPA). (B) IHC staining results for NOP58 in LUAD tissues and adjacent non-tumor tissues (clinical tissue microarray), with the corresponding scatter diagram. Statistical significance was determined by paired t-test. LUAD, lung adenocarcinoma; HPA, The Human Protein Atlas; IHC, immunohistochemistry.
High expression and influence of NOP58 on LUAD cells’ migration and invasion
Compared to BEAS-2B, LUAD cells, such as A549 and H1299, expressed NOP58 at a high level (Fig. 7A). Then, we designed two siRNAs that specifically target distinct NOP58 locations. According to qRT-PCR and Western blot data, both si-RNAs displayed good NOP58 knockdown efficiency (Fig. 7B). The CCK-8 and colony-formation assays were then performed to elucidate cell growth. After NOP58 knockdown, the proliferation rates of A549 and H1299 cells were drastically lowered (Fig. 7C and 7D). The Transwell and wound-healing assays demonstrated that NOP58 knockdown alleviated LUAD cell invasion and migration (Fig. 7E and 7 F).

NOP58 was downregulated in LUAD cells and related to proliferation, migration and invasion in vitro. (A) The expression of NOP58 in LUAD cells was confirmed by qRT-PCR and western blot. (B) The efficiency of SIRNAs was verified by qRT-PCR and western blotting. (C, D) Proliferation of LUAD cells was assessed using the CCK-8 assay (C) and colony-formation assay (D). (E) The knockdown of NOP58 inhibited the invasion of LUAD cells in transwell assays. (F) Knockdown of NOP58 inhibited LUAD cell migration in wound healing assays (* P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001). LUAD, lung adenocarcinoma; qRT-PCR, quantitative reverse transcription-PCR; CCK-8, cell counting kit-8.
NOP58 silencing inhibited tumor growth and enhanced anti-tumor immune activity
To test the anti-tumor effect in vivo, C57BL/6 mice were injected with shNOP58 cells. NOP58 silencing significantly inhibited tumor growth in vivo (Fig. 8A). The tumor volume of mice in the shNOP58 group was remarkably smaller than that in the shCtrl group (Fig. 8B). Compared with the shCtrl group, the shNOP58 group mice showed a lower tumor weight (Fig. 8C). As shown in Fig. 8D, the infiltration of CD8+T cells in shNOP58 group tumors was higher than that in the control group tumors. CD86 is highly expressed on the surface of M1 macrophages. CD86+ macrophages had a higher proportion in the tumors of the shNOP58 group (Fig. 8E).

Knockdown of NOP58 inhibited tumor growth in mouse xenograft models. The mouse xenograft model was established to observe the effects of NOP58 knockdown on tumor volume (A, B), weight (C). D Representative flow cytometry of CD8+ T cells in tumors. E Representative flow cytometry of CD86+ cells in tumors. Quantitative analysis of CD86+ macrophages (** P < 0.01; **** P < 0.0001).
Dissuction
Human lung cancer is the most common malignant tumor, which has a tremendous impact on human health14. In the United States, an estimated 236,740 new lung cancer cases are diagnosed every year, causing 130,180 deaths, which is more than the combined number of colon cancer, breast cancer and prostate cancer cases15. The LUAD histological subtype represents about 50% of lung cancers16. Research shows that the occurrence and development of LUAD is very complex, including gene mutation, gene fusion, epigenetic changes and tumor immune escape16. In recent decades, multimodal treatment strategies including radiotherapy, non-invasive surgical resection, and immunotherapy have progressed immensely, but the results in treating LUAD remain unsatisfactory, and LUAD patients’ 5-year survival rate is only 19%17. It is urgently therefore to find more efficient molecular pathology indicators for diagnosis and prognosis prediction in LUAD.
NOP58 has gradually received attention in recent years for its role in tumors. Due to the paucity of research on the NOP58 gene in cancer, we conducted an exhaustive bioinformatics investigation to discover its biological function and possible regulatory route. It has been found that some lncRNAs interact with the core component of snoRNPs/NOP58, altering NOP58 recruitment, mRNA stability, and consequently tumor growth10,11,12. Despite a review of the scientific literature, we were unable to discover any studies that examined NOP58 in the setting of total malignancies. In this investigation, its expression profile, predictive and prognostic importance, possible roles, interaction network, and connection with immune subset infiltration levels in the human pancancer were identified.
Using three independent datasets (ONCOMINE, TCGA, and GTEx), we initially examined the amount of NOP58 transcription in diverse malignancies. NOP58 was highly expressed in breast, colorectal, gastric, and lung malignancies. Similarly, significant NOP58 expression was observed in LUAD tissues utilizing the HPA dataset and the TMA. It appears that NOP58 might have a crucial function in the development of tumors. Moreover, NOP58 had certain reference values for the diagnosis and prognosis of multiple cancers, which substantially supported its use as a biomarker for oncogenes. Figure 4E indicates a significant difference in NOP58 expression between patients with CR (complete remission) and PD (disease progression), suggesting a potential association of NOP58 with treatment response and disease progression. However, NOP58 expression did not exhibit a significant difference between patients with PR (partial remission) and SD (stable disease), which may be due to a smaller sample size in the PR and SD group. In fact, the number of patients in each group was 67 (CR), 1 (PR), 2 (SD), and 6 (PD), respectively. In addition, the data on NOP58-binding components and expression-associated genes from all tumors for enrichment analyses series were combined; the results suggested that “ribosome biogenesis in eukaryotes”, “spliceosome”, “cell cycle”, and “DNA replication” might be linked with NOP58’s impact on tumor pathogenesis. We verified the protein expression of NOP58 through TMA and HPA database and the data showed that NOP58 protein levels were elevated in LUAD tissues. NOP58 knockdown suppressed the proliferation of LUAD cells, as demonstrated by additional analyses, and it was related to migration and invasion in vitro, and inhibited tumor growth, which indicated its oncogenic driving role in LUAD.
In cancer, malignant cells interact in intricate ways with their tumor microenvironment (TME). Among the immune cells invading the lung TME18,19are T lymphocytes, B cells, DCs, macrophages, and NKs20. In lung cancer, the TME is determined by the percentage of infiltrating immune cells18. LUAD development and progression are linked with tumor-infiltrating immune cells, according to the previous study19. Immunocyte type and level are important prognostic factors as well as immunotherapy response factors. This research demonstrated that the amount of immune infiltration of LUAD was linked with the expression of NOP58. In addition, NOP58 expression was favorably linked with the infiltration of Th2 cells and negatively correlated with the infiltration of cytotoxic cells, CD8 T cells, and Th1 cells. NOP58 might have a critical regulatory function in the immune TME and LUAD development, according to our results. The helper T lymphocytes (Th) are mainly divided into several subsets, such as Th1, Th2, and Th17. Th1 and Th2 respectively dominate the cellular and humoral immunity of the body. When the expression of Th1 is down regulated and the expression of Th2 is up regulated, which means that the balance of Th1/Th2 is shifted, and the immune microenvironment is unbalanced. The body is in an immunosuppressive state, and tumor cells escape from immune surveillance, which promotes the tumors growth and metastasis21,22. The results of this study found that CD8+T cells and CD86+macrophages infiltrated a high proportion of tumor tissues in NOP58 knockdown mice, suggesting that NOP58 may exert carcinogenic effects by regulating the tumor immune environment.
Immune cell migration is facilitated by chemokines and chemokine receptors, which are necessary to mount and then deliver an efficient antitumor immune response. As well, they contribute to the development of pro-tumorigenic microenvironments by generating and recruiting immune cells23. We identified that the levels of NOP58 in LUAD was negatively associated with CCL14, CX3CR1, CCR1, CCR2 and CCR6. The general immune function of CCL14 and CCR1 is participating in innate immunity and adaptive immunity. Meanwhile, CCL14 plays a role in chemokine transcytosis and chemokine scavenging. CCR1-CCL4 mediates the recruitment of Cdc1s. CX3CR1 is the specific signature of CD8 + T cells which is linked with effective anti-tumor immunity24. The decreased expression of these cytokines and their receptors might be related to the insufficient anti-tumor immunity. Additionally, NOP58 expression in LUAD was negatively correlated with CSF1R, HAVCR2, TGFB1, TMEM173, C10orf54 and CD28 expression. Thus, NOP58, which is associated with the aforementioned immune molecules, might furnish as a efficient immunotherapeutic target.
It is indisputable that this study has some limitations. Most of the results of this investigation came from the bioinformatics assessment of the public database. Although we used the clinical TMA to verify the protein level and identified the functional phenotype like proliferation, migration and invasion in vitro by knocking down NOP58 in LUAD cell lines, and further validated NOP58’s carcinogenic effects through in vivo experiments, the in-depth molecular biological process of NOP58 participating in the occurrence and development of LUAD still needs further experimental verification.
Conclusion
Our research showed features of NOP58 in LUAD, including expression profile, genetic mutations, survival prognosis, tumor immune microenvironment, and signaling route. NOP58 might impact the development of LUAD by modulating immunological activity. Moreover, NOP58 had precise reference values for the diagnosis and prognosis of LUAD. The biomarker might serve as a new prognostic factor for patients with LUAD.
Materials and methods
Analysis of gene and protein expressions
An online microarray repository, the Oncomine website (https://www.oncomine.org/) covers 715 datasets and 86,733 samples of different kinds of cancer25. Oncomine might be used to assess biomedical research data retrieval, processing, and display. Using Oncomine analytic techniques, NOP58 mRNA levels were compared between various cancer types and normal tissues. The set thresholds were: P less than 0.001, Fold change greater than 1.5, Gene rank within the top 10%, mRNA data type. In addition, NOP58 mRNA expression level comparisons among cancer tissues with normal tissues of GTEx combined with TCGA, as well as tumor tissue comparisons alongside corresponding paracancer tissues of TCGA, were conducted by the R programming language. And then statistical data analysis and visualizations were generated with “ggplot2” R packages. Moreover, with the GEPIA2’s “Pathological Stage Plot” module (http://gepia2.cancer-pku.cn/), we constructed NOP58 expression violin plots for various stages of pathology (stages I through IV) for all TCGA tumors.
Using UALCAN (http://ualcan.path.uab.edu/analysis-prot.html), an interactive platform to assess the data of cancer genes, we evaluated protein expressions in the CPTAC dataset26. The levels of phosphoprotein (NP 057018.1) and total protein of NOP58 were measured in primary cancers (colon cancer, ovarian cancer, clear cell RCC, breast cancer, LUAD and UCEC) and normal tissues using the term “NOP58.”
Genetic alteration analysis
Researchers could access multidimensional visual data using cBioPortal (http://www.cbioportal.org/), which contains TCGA acquired tumor gene data. We utilized the data of studies from TCGA Pan-Cancer Atlas Studies and mixed pan-cancer studies to answer questions about the genetic alterations of NOP58. The “Cancer Types Summary” module provided the frequency of alteration, type of mutation, and copy number alteration (CNA) data for all TCGA tumors. In addition, we examined the co-occurrence patterns of gene mutations between NOP58 signatures alongside other proteins among 10,953 participants indicated in TCGA Pan-Cancer Atlas Studies via the cBioPortal27. Using a log ratio greater than 5, q-value less than 10−10, and p-value less than 10−10, we performed gene alteration co-occurrence analysis.
Analysis of NOP58-related gene enrichment
Initially, we perused the STRING database (https://string-db.org/) through queries “NOP58” and “Homo sapiens”. There were four primary parameters established: minimum interaction score (“high confidence (0.900)”), network edges’ meaning (“evidence”), maximum interactors to show (“no more than 40 interactors” in the 1st shell), active interaction sources (“experiments”). We got the NOP58-binding proteins as determined by experiments. Then, we analyzed GEPIA2’s “Similar Gene Detection” module using data from all normal tissues and TCGA cancers to find the top 100 NOP58-correlated targeting genes Additionally, GEPIA2’s “correlation analysis module” was utilized for conducting a pairwise gene Pearson correlation study of NOP58 alongside chosen genes. In addition, the TIMER2 module gave heatmap data for selected genes, including a partial correlation (cor) and P-value for pure-adjusted Spearman’s rank correlation. We compared genes that bind to NOP58 and those that interact with it using Jvenn28, an interactive viewer for Venn diagrams. After the two data sets were joined, the KEGG pathway (www.kegg.jp/kegg/kegg1.html)29and GO enrichment assessments were carried out. Visualizations were created via the “clusterprofiler” and “org.hs.eg.db” R packages. Probability values below 0.05 were regarded as statistically significant.
Survival prognosis analysis with Kaplan-Meier plotter
We assessed NOP58 predictive values via an open database Kaplan-Meier plotter, which incorporates clinical data, including prognosis, tumor genes’ mRNA levels, and patient survival status and period30. In this work, median NOP58 gene expression data from cancer patients were utilized to categorize them into low- and high- expression cohorts, principally using data from TCGA (RNA-seq) and Gene Expression Omnibus (GEO) (microarray). We used Kaplan-Meier survival curves to examine the relationship of NOP58 levels with OS and RFS in 21 types of cancers. Hazard ratios (HRs) were calculated using log-rank P-values and 95% confidence intervals.
Diagnostic value analysis
NOP58’s diagnostic value for pan-cancers was assessed via ROC. The range of area under the ROC curve was from 0.5 to 1. The diagnostic effect is best when the AUC is near 1. Low accuracy can be obtained with AUCs between 0.5 and 0.7, moderate accuracy can be obtained with AUCs between 0.7 and 0.9.
Correlational analysis of gene expressions with therapeutic responses
We analyzed relationships between NOP58 transcriptome levels and therapy among patients with glioblastoma, breast, ovarian, or colorectal cancers using the ROC plotter service (http://www.rocplot.org/) to determine the influence of NOP58 on therapeutic responses. The ROC plotter can identify gene expression correlates with therapy response via transcriptomic data obtained from 3,104 breast cancer patients and 2,369 ovarian cancer patients31.
Using Tumor Immune Dysfunction and Exclusion (TIDE) (http://tide.dfci.harvard.edu/), an online prediction tool, we first estimated the responder rate for samples with greater or lower NOP58 expression. TIDE examines the probability of tumor immune escape using gene expression data, notably in melanoma and NSCLC32. Only the top 50 or bottom 50 OLR expression data instances were selected from the 284 samples due to tool constraints. To study the particulars of the anticipated therapeutic impact, we calculated predictive indicators and reorganized the results into a figure that was easier to read.
NOP58 expression and various clinical attributes in LUAD
We utilized box plots to illustrate NOP58 expression levels in LUAD patients with diverse clinical features. The TCGA database was used to get RNA-seq and clinical information in level 3 HTSeq-fragments/kilobase/million formats and convert them to transcripts per million reads (TPM). Following log2 conversion, the data were examined. These two data cohorts were identified by the Wilcoxon rank sum test. The p-value of < 0.05 was termed statistically significant.
Univariate and multivariate cox regression analyses in LUAD
NOP58 and clinical attributes were analyzed for the prognostic values of NOP58 in PFI, DSS, and OS in LUAD. “survival” package was used for statistical analysis.
Immune infiltration analysis
The online server called TIMER2 (https://portal.gdc.cancer.gov/) was utilized33 to study and assess the association between NOP58 levels and immune infiltrates in all TCGA tumors. Through TIMER, we investigated gene expression levels in both malignant and normal tissues from various cancers. Deconvolution of gene expression patterns from LUAD samples in the TCGA datasets was utilized to estimate the number of immune cells invading tumors (TIICs). To determine P-values, a purity-adjusted Spearman’s rank correlation test was used. Using violin plots and scatter plots, the data was represented graphically.
TISIDB database analysis
TISIDB (http://cis.hku.hk/TISIDB/) is essential for identifying the interactions of tumor and immune system34. To understand the immunological connection of NOP58 in cancer, we analyzed and evaluated the association between NOP58 expression against immune checkpoint gene levels using TISIDB’s “Immunomodulator” module. Using the “chemokine” module, we assessed the levels of chemokine and chemokine receptor expression in TIICs to examine links between NOP58 and chemokine and chemokine receptor expressions.
Immunohistochemistry staining of NOP58
HPA (https://www.proteinatlas.org) is a publicly available database of immunohistochemistry pictures analyzing protein expression patterns in roughly 20 common cancers and normal tissues35. It compared immunohistochemical pictures of LUAD specimens with normal tissue samples to estimate the degree of NOP58 protein expression in each.
On a human LUAD TMA (Cat# AF-LucSur2202, AiFang biological, Changsha, China), the expression of NOP58 was evaluated by immunohistochemistry. The AiFang Biological Ethics Committee accepted the use of human volunteers in this investigation. Immunohistochemistry (IHC) staining was conducted on TMA blocks containing a total of 160 cases (80 cancer tissue samples and 80 paired adjacent tissues of cancer). The anti-NOP58 antibody (1:1000 dilution, cat# ab155969) was obtained from Abcam (Cambridge, United States). The study using the tissue microarray was approved by the Life Sciences Ethics Committee of Changsha Yaxiang Biotechnology Co., LTD. The query code is 3XLN6BC700EGHE. We conducted the study by following the Declaration of Helsinki. Informed consent was obtained from all subjects and/or their legal guardian(s).
Cell culture and transfection
The Chinese Academy of Sciences (Shanghai, China) provided normal human lung epithelial (BEAS-2B) and human lung cancer (A549 and H1299) cell lines. We maintained these cells in Roswell Park Memorial Institute 1640 medium (RPMI 1640; 10% FBS) with suitable conditions (37.5 °C; 5% CO2). KeyGEN BioTECH (Nanjing, China) provided siRNAs (si-NC, si-NOP58-1, and si-NOP58-2) for transfection using Lipofectamine 3000 (Invitrogen, USA). siRNA sequences are provided in Table S8.
Quantitative reverse transcription-PCR (qRT-PCR)
Using the reverse transcription kit (Takara, Japan), the total RNA from cells inoculated with Trizol (Takara, Japan) was reversely transcribed. We used the SYBR Green PCR kit (Takara, Japan) for NOP58 qRT-PCR analysis. 2−△△Ct method was used to normalize NOP58 expression to GAPDH expression. In Table S9, primer sequences were listed.
Western blot assay (WB)
After the collection of cell and tissue samples, the BCA Protein Assay Kit (Beyotime, China) was utilized for protein quantification in cells and tissues. First, protein isolation was performed via sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Then, 5% milk was then used to inhibit proteins on the polyvinyl fluoride membrane. The membrane was then overnight treated with specified primary antibodies (NOP58, Abcam; GAPDH, Abcam). After incubation with secondary antibodies, we utilized the ECL Western Blotting Substrate (Solarbio, China) for identifying protein blots.
Cell counting kit-8 (CCK8) assay
For each experimental group, logarithmic growth phase cells were trypsinized, resuspended in a full medium, then cultivated overnight. Cell proliferation was determined using the CCK-8 reagent (Abcam) after 24, 48, 72, and 96 h, per the manufacturer’s instructions. A microplate reader (Molecular Devices, Rockford, IL, USA) was utilized to elucidate the optical density at 450 nm.
Colony-formation assay
For nine days, 500 LUAD cells were grown on six-well plates. The cell colonies were fixed in 10% ethanol for 5 min and stained with crystal violet (Beyotime, China). Colonies of cells were photographed and tallied.
Wound-healing assay
Pipette tips were used for drawing the cell layer surface after culture of A549 and H1299 cells in plates for 24 h. We used a microscope to capture images of the injury area at 0 h, and then took the images again after 24 h. An injury area’s distance was measured at 24 h, and after normalizing distance at 0 h, a relative migration rate was calculated.
Transwell assay
Transwell experiment was conducted in a matrigel-filled transwell chamber. The top chamber layer was injected with a mixture of serum-free media and cells. On the outside of the transwell chamber, medium was present. Transwell chambers were stained with 4% paraformaldehyde and 0.1% crystal violet (Solarbio, China) after 48 h of cell growth. In addition, the cells at the chamber bottom were counted.
Tumor formation assay in C57BL/6 mice
Using simple randomization, C57BL/6 female mice (Tianjin Gecon Med Tech Co., Ltd, Tianjing, China) aged 6–7 weeks were randomly divided into two groups to generate a subcutaneous tumor mouse model. A549 cells transfected with shCtrl and shNOP58, washed and resuspended in PBS, were harvested. 5 × 106 cells in 100 µl PBS were injected into the right axilla of each mouse. The following formula was used to determine tumor volume: (length × width2)/2. Once the tumor size reaches 2000 mm, the mice were euthanized through cervical dislocation, following the induction of deep anesthesia (pentobarbital sodium). All animal studies were approved by the Cell Ecology Haihe Laboratory Experimental Animal Welfare Ethics Committee (HH-DWLL-YW2024004-1). This study was performed in accordance with the Animal Ethical and Welfare Committee (AEWC) and were in accordance with the ARRIVE guidelines.
Statistical analysis
In order to analyze the data, SPSS 22.0 (IBM, USA) was used. Student’s t-tests, one or two-way variance analysis were performed. The Turkey method or Scheffe method was used for pairwise comparisons in multiple groups. As a statistically significant result, P < 0.05 was considered. All the data was depicted as mean ± SD.
Data availability
The datasets analyzed in this study are available in the following open access repositories: Oncomine, https://www.oncomine.org/; TCGA, https://portal.gdc.cancer.gov/; GTEx, www.gtexportal.org/; TIMER 2.0, http://timer.comp-genomics.org/; TIDE, http://tide.dfci.harvard.edu/; cBioPortal, http://www.cbioportal.org; GEPIA2, http://gepia2.cancer-pku.cn/; UALCAN, http://ualcan.path.uab.edu/analysis-prot.html; STRING database, https://string-db.org/; ROC plotter service, http://www.rocplot.org/; HPA, https://www.proteinatlas.org. The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Cancer World Health Organization https://www.who.int/news-room/factsheets/detail/cancer (2018). Accessed 2023-06-01
Stella, G. M., Luisetti, M. & Pozzi, E. Primary lung adenocarcinoma: characteristics by smoking habit and sex. Eur. Respir J. 39 (4), 1051–1052 (2012).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 68 (6), 394–424 (2018).
Barta, J. A., Powell, C. A. & Wisnivesky, J. P. Global epidemiology of Lung Cancer. Ann Glob Health 85 (1), 8 (2019).
LymanSK, Gerace, L. & Baserga, S. J. Human Nop5/Nop58 is a component common to the box C/D small nucleolar ribonucleoproteins. RNA (New York NY). 5 (12), 1597–1604 (1999).
Boisvert, F. M., van Koningsbruggen, S., Navascués, J. & Lamond, A. I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 8 (7), 574–585 (2007).
Liang, J. et al. Small nucleolar RNAs: insight into their function in Cancer. Front. Oncol. 9, 587 (2019).
Wang, J., Huang, R., Huang, Y., Chen, Y. & Chen, F. Overexpression of NOP58 as a prognostic marker in Hepatocellular Carcinoma: a TCGA Data-based analysis. Adv. Therapy. 38 (6), 3342–3361 (2021).
Shen, Z., Liu, S., Liu, J., Liu, J. & Yao, C. Weighted Gene Co-expression Network Analysis and Treatment Strategies of Tumor Recurrence-Associated Hub Genes in Lung Adenocarcinoma. Front. Genet. 12, 756235 (2021).
Wu, H. et al. Long noncoding RNA ZFAS1 promoting small nucleolar RNA-mediated 2’-O-methylation via NOP58 recruitment in colorectal cancer. Mol. Cancer. 19 (1), 95 (2020).
Qi, L., Sun, B., Yang, B. & Lu, S. PGM5P3-AS1 regulates MAP1LC3C to promote cell ferroptosis and thus inhibiting the malignant progression of triple-negative breast cancer. Breast Cancer Res. Treat. 193 (2), 305–318 (2022).
He, J. & Yu, J. Long noncoding RNA FAM83A-AS1 facilitates hepatocellular carcinoma progression by binding with NOP58 to enhance the mRNA stability of FAM83A. Biosci. Rep. 39 (11), BSR20192550 (2019).
Dall’Olio, F. G. et al. Tumour burden and efficacy of immune-checkpoint inhibitors. Nat. Reviews Clin. Oncol. 19 (2), 75–90 (2022).
Cancer today. Gco.iarc.fr. (2022). https://gco.iarc.fr/today/home (2022). Accessed 2023-06-11
Cancer types. Natl. Cancer Inst. https://www.cancer.gov/types/lung (2023). Accessed 2023-06-01
Caso, R. et al. The underlying Tumor Genomics of predominant histologic subtypes in Lung Adenocarcinoma. J. Thorac. Oncol.: Off. Publ. Int. Association Study Lung Cancer. 15 (12), 1844–1856 (2020).
Hirsch, F. R. et al. Lung cancer: current therapies and new targeted treatments. Lancet. 389 (10066), 299–311 (2017).
Zheng, X., Hu, Y. & Yao, C. The paradoxical role of tumor-infiltrating immune cells in lung cancer. Intractable rare Dis. Res. 6 (4), 234–241 (2017).
Bremnes, R. M. et al. The role of Tumor-infiltrating lymphocytes in Development, Progression, and prognosis of Non-small Cell Lung Cancer. J. Thorac. Oncology: Off. Publ. Int. Association Study Lung Cancer. 11 (6), 789–800 (2016).
Domingues, P. et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain. Behav. Immun. 53, 1–15 (2016).
Lin, W. et al. Imbalance of Th1/Th2 and Th17/Treg during the development of uterine cervical cancer. Int. J. Clin. Exp. Pathol. 12 (9), 3604–3612 (2019).
Bing, X., Xuelei, L., Wanwei, D., Linlang, L. & Keyan, C. EGCG maintains Th1/Th2 balance and mitigates Ulcerative Colitis Induced by Dextran Sulfate Sodium through TLR4/MyD88/NF-κB signaling pathway in rats. Can. J. Gastroenterol. Hepatol. 2017, 3057268 (2017).
Anichini, A., Perotti, V. E., Sgambelluri, F. & Mortarini, R. Immune escape mechanisms in non small cell Lung Cancer. Cancers 12 (12). (2020).
Ozga, A. J., Chow, M. T. & Luster, A. D. Chemokines and the immune response to cancer. Immunity. 54 (5), 859–874 (2021).
Rhodes, D. R. et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia (New York NY). 6 (1), 1–6 (2004).
Chandrashekar, D. S., Bashel, B., Balasubramanya, S. A. H., Creighton, C. J., Ponce-Rodriguez, I., Chakravarthi, B., et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia (New York, NY). 19 (8), 649 − 658 (2017).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2 (5), 401–404 (2012).
Bardou, P., Mariette, J., Escudié, F., Djemiel, C. & Klopp, C. Jvenn: an interactive Venn diagram viewer. BMC Bioinform. 15 (1), 293 (2014).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51 (D1), D587–d92 (2023).
Lánczky, A. & Győrffy, B. Web-based Survival Analysis Tool tailored for Medical Research (KMplot): development and implementation. J. Med. Internet. Res. 23 (7), e27633 (2021).
Fekete, J. T. & Győrffy, B. ROCplot.org: validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int. J. Cancer. 145 (11), 3140–3151 (2019).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24 (10), 1550–1558 (2018).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48 (W1), W509–w14 (2020).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics: J. Integr. Biology. 16 (5), 284–287 (2012).
Navani, S. Manual evaluation of tissue microarrays in a high-throughput research project: the contribution of Indian surgical pathology to the human protein atlas (HPA) project. Proteomics. 16 (8), 1266–1270 (2016).
Acknowledgements
We acknowledge TCGA and other public databases for providing their platforms and contributors for uploading their meaningful datasets. The graph abstract was created by Figdraw (www.figdraw.com). This work was supported by 2024 Zhejiang Province Medical and Health Science and Technology Plan (2024KY753) and 2024 Zhejiang Province Traditional Chinese Medicine Science and Technology Plan (2024ZL256).
Author information
Authors and Affiliations
Contributions
QSS and CRD designed the project, analyzed the results, and wrote the manuscript. LHF performed the related experiments. ZM, ZL and DYL performed the statistical analysis. YXM and WWS reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qian, S., Liu, H., Zhang, M. et al. Pan-cancer landscape analysis of NOP58 and its oncogenic driving role in lung adenocarcinoma. Sci Rep 14, 27583 (2024). https://doi.org/10.1038/s41598-024-77500-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-77500-9
