
Abstract
In the realm of epigenetic regulation, Protein arginine deiminase 2 (PAD2) stands out as a therapeutic target due to its significant role in neurological disorders, rheumatoid arthritis (RA), multiple sclerosis (MS), and various cancers. To date, no in silico studies have focused on PAD2 for lead compound identification. Therefore, we conducted structure-based pharmacophore modeling, virtual screening, molecular docking, molecular dynamics (MD) simulations, and essential dynamics studies using PCA and free energy landscape analyses to identify repurposed drugs and selective inhibitors against PAD2. The best pharmacophore model, ‘Pharm_01,’ had a selectivity score of 10.485 and an excellent ROC curve quality of 0.972. Pharm1 consisted of three hydrogen bond donors (HBD) and two hydrophobic (Hy) features (DDDHH). A virtual screening of about 9.2 million compounds yielded 2575 hits using a fit value threshold of 2.5 and drug-likeness criteria. Molecular docking identified the top ten molecules, which were verified using MD simulations. Stability was verified using MM-PBSA studies, whereas conformational differences were investigated using PCA and free energy landscape analyses. Two hits (Leads 1 and 2) from the DrugBank dataset showed promise for repurposing as PAD2 inhibitors, while one hit compound (Lead 8) from the ZINC database emerged as a novel PAD2 inhibitor. These findings indicate that the discovered compounds may be potent PAD2 inhibitors, necessitating additional preclinical and clinical research to produce viable treatments for cancer and neurological disorders.
Similar content being viewed by others
Introduction
Protein arginine citrullination has been highlighted as a crucial post-translational modification (PTM) ever since the discovery of arginine residue citrullination1,2,3. Dysregulation of citrullination is associated with various pathogenic processes, including Alzheimer’s disease, rheumatoid arthritis (RA), ulcerative colitis (UC), lupus, multiple sclerosis (MS), and several cancers4,5. Enzymes belonging to the protein arginine deiminase (PAD) family catalyze the citrullination of arginine, with five known PADs (1–4, and 6) identified in mammals6,7,8. This modification transforms positively charged arginine residues in substrate proteins or receptors into nonstandard neutrally charged citrulline, thereby influencing protein structure and inducing changes that may lead to protein denaturation9. PADs are ubiquitously distributed across different tissues and contribute to various diseases10. All PADs (denoted by the gene symbol PADI) possess highly conserved calcium-binding residues, except for PAD6, which lacks some of these sites and features a distinct catalytic cysteine PAD611. The human PADI genes are clustered on the short arm of chromosome 1, proximal to the telomere, covering an approximately 334.7 kb region (1p36.1). These genes are sequentially arranged in a p-telomere order, with the order being p-telomere → PADi2 → 1 → 3 → 4 → 6. Despite substantial similarity (around 50–60%) in the coding nucleotide sequences of all PADI genes, diverse regulatory mechanisms govern their distinct expression patterns8,12.
Among the PADs, PAD2 exhibits a broader expression pattern, being present in various organs and tissues, including the epidermis, bone marrow, brain, kidney, skeletal muscles, breast, uterus, spleen, and pancreas13. While primarily cytoplasmic, a fraction of PAD2 in human mammary epithelial cells may translocate to the nucleus for histone citrullination. The regulation of PAD2 expression involves both protein translation and mRNA splicing. Notably, PAD2 plays crucial roles in neurological disorders, rheumatoid arthritis (RA), multiple sclerosis (MS), and various cancers by citrullinating histone and nonhistone proteins (such as myelin basic protein, actin, and vimentin). These citrullinated proteins control apoptosis, autophagy, epithelial-to-mesenchymal transition (EMT), angiogenesis, and other cellular processes14,15,16,17. PAD2’s functional site comprises three domains: two immunoglobulin-like domains (IgG1; residues 1-115 and IgG2; residues 116–295) and a well-characterized C-terminal ___domain (residues 296–665). Like other PADs, PAD2’s overall activity is regulated by the calcium switch (Ca3-5 sites), converting the protein from its apo form to its holo form. This transition facilitates the protection of the active site (Arg347) and the proper placement of the catalytic cysteine (Cys647). The ordered binding of six calcium ions (Ca1-6) suggests that PAD2 exhibits a higher affinity for calcium binding compared to PAD4, which lacks the recently discovered Ca6 site involved in epithelial-to-mesenchymal transition (EMT), angiogenesis, and other cellular processes11.
The pivotal role of PAD2 has attracted considerable attention across various diseases. McElwee et al. reported a noteworthy upregulation of PADI2 in the MCF10AT model, impacting both mRNA and protein levels18. Zhang et al. linked PAD2 to histone H3R26 citrullination at ER targets, thereby influencing gene transcription16. In the context of breast cancer, Wang et al. identified the impact of PAD2 on BINC3, ACSL4, and CA9, promoting aberrant cell invasion and lipid metabolism19. Horibata et al. demonstrated PAD2’s involvement in cytoskeletal regulation and mammary gland hyperbranching20. Li et al. associated PAD2 with tamoxifen resistance in breast cancer, proposing a synergistic drug regimen BC21. In castration-resistant prostate cancer, the upregulation of PADI2 activates androgen receptor signaling, contributing to cancer progression22. Furthermore, PADI2’s tumorigenic role manifested in endometrial23 and lung cancer carcinoma24, suggesting its potential as a therapeutic target. Collectively, these findings underscore the diverse implications of PAD2 in the development and progression of several cancers.
Numerous studies have delved into the design of PAD inhibitors to elucidate the involvement of PAD in diverse diseases. While reversible PAD inhibitors such as paclitaxel, minocycline, and streptomycin exist, their efficacy remains suboptimal, particularly for acetamidine halogen-based inhibitors25. Consequently, Cl-amidine analogs26, exemplified by O-F-amidine and O-Cl-amidine, demonstrate enhanced efficacy and selectivity, with O-F-amidine exhibiting a 65-fold increase in effectiveness against PADs compared to F-amidine)27,28. Noteworthy PAD2-specific inhibitors like AFM30a, AFM41a, and AFM49a display therapeutic potential. Challenges persist, encompassing limited effectiveness in cell-based assays for existing PAD inhibitors and the absence of clinically approved PAD2-selective inhibitors29. Computational strategies, including structure-based virtual screening, have been employed for drug discovery. Several studies have identified potential PAD4 inhibitors, with molecules exhibiting IC50 values ranging between 1.54 and 3.0 mM30. While certain compounds31 show promise against PAD4, investigations utilizing in silico methods for PAD2 screening are currently lacking. Given the pivotal role of PAD2 in human diseases, our study employs structure-based pharmacophore modeling, virtual screening, molecular docking, covalent docking, MD simulation, and MM-PBSA analyses to develop selective PAD2 inhibitors.
Materials and methods
Computational and software details
In this study, Accelrys Discovery Studio 4.0, Cresset’s Flare 5.0, and Biovia Discovery Studio 202032 were employed for various computational tasks, including structure-based pharmacophore modeling, virtual screening, protein preparation, molecular docking, and visualization. All these computational processes were carried out on a Dell Xeon workstation. Molecular dynamics (MD) simulations and MM-PBSA studies were executed using GROMACS 2019.233. The calculations were performed on an HP workstation and the CDAC-BRAF Pune server. The analysis of MD trajectory graphs was conducted using Xmgrace software.
Dataset selection and preparation
Due to the absence of a PAD2 structure with an inhibitor in the Protein Data Bank, we capitalized on the substantial structural similarity and sequence identity shared between PAD4 and PAD2. Notably, critical residues projecting into the binding site exhibited conservative substitution or identity. To elucidate the ligand binding features with PAD2, we utilized the structure of human PAD4 (PDB ID: 5N0Y) as a reference29. This reference structure was superimposed with PAD2 (PDB ID: 4N2C)11 to delineate the binding site. Subsequently, we performed molecular docking using the “CDOCKER module” in Discovery Studio (DS) 4.0 of the co-crystallized PAD4 ligand, “AFM-30a,” with PAD2, given that AFM-30a serves as a selective inhibitor for PAD2.
Before commencing pharmacophore modeling, the target structure underwent necessary preparations using the “protein preparation module” in DS4.034.
Structure-based pharmacophore model generation
The Structure-Based Pharmacophore Model (SBPM) offers insights into the intermolecular interactions that stabilize protein-inhibitor complexes. Using the Receptor-Ligand pharmacophore generation tool in DS 4.0, pharmacophore hypotheses were developed employing the Genetic Function Approximation (GFA) technique35. Some default values were adjusted for specific distances: Max_Charge distance was extended to 6 Å from 5.6 Å, Max_Hydrogen bond distance was modified to 3.7 Å from 3.0 Å, Max_Hydrophobic distance was set to 6.4 Å from 5.5 Å, and Min_Interfeature distance was reduced to 1.5 Å from 2.0 Å.
The resulting pharmacophore model, derived from receptor-ligand interactions, encompassed crucial features such as Hydrogen bond donor (HBD), Hydrogen bond acceptor (HBA), Hydrophobic (HY), Positive ionizable (PI), Negative ionizable, and Ring Aromatic (RA).
Pharmacophore validation
To evaluate the capability of the pharmacophore in distinguishing PAD2 inhibitors, we utilized the decoy set method, dividing the datasets into 40 actives and 1000 decoy set molecules, representing positive and negative controls, respectively36. A robust pharmacophore model is expected to effectively discriminate between active and inactive compound datasets, recognizing pharmacophoric features from the former while rejecting those from the latter. Additionally, Receiver Operating Characteristics (ROC) curves were also computed to validate our pharmacophore models37.
Database preparation
In this investigation, we harnessed four chemical databases, encompassing a total of 9,191,288 compounds, for virtual screening against the designated pharmacophore. These databases comprised the ZINC15 database (https://zinc15.docking.org)38, Otava’s collections (drug-like green, lead-like, and anticancer library), DrugBank for drug repurposing (https://go.drugbank.com/), and the kinase library from Selleckchem39, with 8,900,485, 270,020, 12,448, and 8,335 compounds, respectively. To handle the considerable number of compounds in the ZINC15 database, the “Build 3D database module” in DS 4.0 was utilized for their integration. As a result, a unified chemical library of 9,191,288 compounds was meticulously prepared.
Furthermore, a covalent chemical library comprising 843 compounds from the Selleckchem database was specifically curated and readied for covalent docking.
Pharmacophore-based virtual screening
Pharmacophore-based virtual screening entails the identification of compounds that align optimally with the pharmacophore, ensuring a comprehensive mapping of all features40. Compounds from the unified chemical library underwent screening against the pharmacophore model to unveil novel scaffolds as potential PAD2 ___domain-selective inhibitors. The screening process was efficiently facilitated using DS 4.0’s “Search 3D Database module” (for ZINC15) and “Screen Library module” (for Otava, DrugBank, and Selleckchem).
Hit compounds were prioritized based on their pharmacophore fit values (≥ 2.5), recognizing that certain approved drugs have been withdrawn from the market due to poor ADMET properties41, we implemented pre-emptive filtering at this stage to conserve time and resources. The selected hit compounds underwent scrutiny against Lipinski and Veber Rules, alongside the ADME protocol in DS 2020. To evaluate potential toxicity, the TopKat® module of DS 202042 was employed, providing an additional refinement layer in the compound selection process.
Molecular docking
High-throughput screening using LibDock
Compounds were prepared using the “Prepare or Filter Ligands module” present in Biovia DS 2020. Following additional preparation and energy minimization using DS 2020, a stringent RMS gradient of 0.001, employing 4000 steps of steepest descent followed by 4000 steps of conjugate gradient, was achieved. High-throughput docking with LibDock43 against PAD2’s tertiary structure (PDB ID: 4N2C) ensued. LibDock efficiently screens both ligands and proteins, pinpointing binding hotspots and ranking poses based on the LibDock score42.
Docking of hits using CDOCKER
The top 10% of compounds (257 hit molecules) identified from the LibDock results were then subjected to molecular docking using the CDOCKER module of DS 2020, which is a CHARMm-based molecular dynamics simulated docking tool44. In this step, ligands were allowed to be flexible while the receptor remained rigid. The predefined binding site of PAD2 (PDB ID:4N2C) was used for molecular docking and a grid sphere of 228.822, -17.8375, and 22.5556 points with a radius of 8.99 A° was set. The following residues were defined within the binding site sphere: Asp346, Arg347, Trp348, Asp351, Arg373, Ser404, Ser407, Thr468, Val469, His471, Asp473, Tyr638, His639, Lys640, Phe641, Leu642, Gly643, and Cys647. The hit compounds with the best docking conformation were chosen based on their interactions with crucial binding site residues and low CDOCKER energy.
To ensure the reliability of the molecular docking results, we applied several standard scoring algorithms, including PLP1, PLP2, LigScore1, LigScore2, PMF, PMF04, Jain, Ludi Energy Estimate-1, Ludi Energy Estimate-2, and Ludi Energy Estimate-3, in Biovia DS 2020 to score each ligand conformation42.
Covalent docking
In the exploration of the covalent inhibitors45 tools, Cresset’s Flare 5.046 was utilized for docking against 843 compounds from Selleckchem’s covalent library. Targeting PAD2 (PDB ID: 4N2C) and utilizing AFM-30a as a control, the docking parameters specified Cys647 for covalent bonding. The post-docking analysis employed the LF-VS score ( Cresset’s Flare 5.0) 47 for ranking and binding energy (Dg) to ascertain the correct binding pose. This method aims to unveil potential covalent inhibitors for PAD2, addressing their selectivity and interaction in the active site region.
Molecular dynamics simulation
A 100 ns molecular dynamics (MD) simulation was performed using the GROMACS 2019.2 package48 with GROMOS96 forcefields49 to assess the protein-ligand complex’s dynamic behavior and stability. We selected the top 10 complexes based on -CDOCKER energy scores from initial docking results for these simulations. These complexes were placed at least 1.0 nm from the edges in a cubic box, solvated using the SPC water model50, and neutralized with sodium and chloride ions. Ligand topologies were generated via the Prodrg server and integrated into the protein topology. The system underwent energy minimization using the steepest descent algorithm for 50,000 steps to resolve steric clashes. LINCS was used to maintain bond length constraints, and the particle mesh Ewald method handled electrostatic calculations51. Following energy minimization, the system was equilibrated under NVT and NPT conditions for 100 ps at 300 K and 1 atm pressure. A 100 ns MD simulation was then conducted with a 2-fs integration step, during which system stability, interactions, and fluctuations were analyzed using various parameters like RMSD, RMSF, RoG, and H-bond analysis42.
To further validate the MD simulation results, binding free energies and residue decomposition analysis were calculated for the selected hits and a control compound (AFM30a) using the MM-PBSA method with the g_mmpbsa tool52. The binding energies were extracted from the last stable 20 ns of the MD simulations, and the ΔGbind was determined using the equation:
ΔGbind.pred = ΔGcomplex – [ΔGRec + ΔGlig].
In the above equation, ∆Gcomplex represents the free energy change in forming the protein-ligand complex, while ∆GRec and ∆Glig are the free energy changes associated with the unbound receptor and ligand, respectively.
Principal component analysis
From MD simulations, Principal component analysis (PCA)53 was used to explore the conformational dynamics of PAD2-inhibitor complexes. PCA focused on Cα atom displacements to assess global motion in complexes with AFM-30a and top hits from MM-PBSA results. The analysis was conducted using the GROMACS 2019.2 package, where a covariance matrix was generated, and the first two principal components (PC1 and PC2) were analyzed for significant collective motions54. Additionally, the free energy landscape was predicted to gain insights into the stability and preferred conformational states of the complexes.
Binding mode analyses
Binding mode analyses, a key approach in drug discovery, describe how a compound’s structural features influence its interactions with a target protein55. In the present study, we performed binding mode analysis to evaluate how the structural properties of the shortlisted lead compounds affect their binding affinity with the PAD2 enzyme. The analysis focused on the lead compounds (Lead 1, Lead 2, Lead 8, and Lead 9) selected from the top hits of the MM-PBSA analysis, based on their binding energy to PAD2, and compared them with the positive control molecule AFM30A. The interactions between these lead compounds and the PAD2 enzyme were thoroughly examined using Biovia DS 2020 to identify critical residues and key interactions.
Results and discussions
Tertiary structure selection and preparation for PAD2 and PAD4 enzymes
In this study, we selected the holo form of Protein Arginine Deaminase 2 (PAD2) based on experimental studies by Slade et al. (PDB ID: 4N2C)11 and the crystal structure of PAD4 co-crystallized with the selective inhibitor AFM-30a (PDB ID: 5N0Y)29. We extracted binding site information based on PAD4’s crystal structure. Structural studies revealed a highly conserved active site architecture in both PAD2 and PAD4 despite substrate specificity and activity differences. The catalytic site, accommodating the substrate arginine residue (Arg373), features conserved cysteine (Cys647) and histidine residues (His471) at the bottom of the pocket (Fig. 1a–c).

Structural and sequence similarities between PAD2 and PAD4, a Superimposed protein structures of PAD2 (green) and PAD4 (yellow) for the identification of the active site (red) of PAD2 based on the catalytic site information of PAD4, b 3D interactions of “AFM-30a” highlighted in red color with PAD2 (green) and PAD4 (yellow)in superimposed form, c Sequence similarity between PAD2 and PAD4. In the active site, important catalytic site residues encircled with red color were conserved in both isoforms.
The crystal structures were prepared using DS 4.0’s protein preparation module to ensure correct 3D structures. The preparation protocol included adding missing atoms, removing water and heteroatoms, error correction, protonation state assignment, removing alternative conformation, and hydrogen bonding network optimization56. Post-preparation, AFM-30a was docked into PAD2’s active site using the CDOCKER module of DS 4.0. The docked complex of PAD2 with AFM-30a was then utilized to create 3D structure-based pharmacophore models, facilitating a comprehensive exploration of the active site for potential PAD2 inhibitors.
Structure-based pharmacophore model generation and validation
In this study, we employed pharmacophore modeling, a computational approach aimed at identifying essential chemical and structural features crucial for the interaction of ligands with a biological target57. To pinpoint the binding site for molecular docking on PAD2, we selected the first conformation from the generated ten poses after docking with CDOCKER, guided by critical catalytic activity interactions and the highest -CDOCKER interaction energy score (-42.1515 kcal/mol).
The interactions between AFM-30a and PAD2’s active site amino acid residues are vividly illustrated in Fig. 2a. The interaction profile of AFM-30a (control compound) with PAD2 reveals a complex network of binding interactions. The fluoroacetamidine group of AFM-30a forms hydrogen bonds with Asp351 and Leu642 while also engaging in hydrophobic interactions with Trp348, Val469, and Cys647. The indole ring of AFM-30a further stabilizes through a hydrogen bond with Trp348 and hydrophobic interactions with Phe641. Additionally, the benzimidazole group is also positioned towards Thr468 and Arg582, establishing van der Waals interactions. Other crucial van der Waals interactions involve residues Gly345, Asp346, Arg347, Asn376, Arg373, Ser404, Ser407, Arg441, His471, Asp473, and Gly643. This intricate network of hydrogen bonds, hydrophobic interactions, and van der Waals forces underscores the binding stability and specificity of AFM-30a with PAD2.

Structure-based pharmacophore model generation and validation. (a) 2D interaction analysis of human PAD2 with AFM-30a, (b) Pharmacophoric features in the presence of receptor-ligand interactions, (c) Best pharmacophore model (DDDHH), (d) ROC curve displaying the quality of the model.
Utilizing this conformation, we generated a structure-based pharmacophore model to scrutinize the optimal model for AFM-30a binding to PAD2. The most favorable pharmacophore model (Pharmacophore_01), designated as Pharm_01 out of the 10 generated, showcased five pharmacophoric features—comprising three Hydrogen Bond Donor (HBD) and two Hydrophobic (Hy) features (DDDHH), yielding a selectivity score of 10.485 (Fig. 2b; Table 1). Notably, incorporating excluded volumes within a 4Å radius of the ligand during pharmacophore generation heightened accuracy by confining potential ligand choices to areas aligning with active site amino acid residues.
For clarity in presenting the pharmacophore model, excluded volumes are intentionally omitted in Fig. 2c as their inclusion might overshadow the discernibility of pharmacophoric features. The validation of the pharmacophore model meticulously considered both the quantity and diversity of features, ensuring its reliability and high quality for subsequent analyses. This comprehensive approach emphasizes the precision of the structure-based pharmacophore model for PAD2 interaction.
Pharmacophore validation using external active set and decoy set of inhibitors
To authenticate the pharmacophore model generated in the preceding step, the well-established decoy set method was employed—a technique renowned for its efficacy in validation58. This method filters an external dataset for active and decoy compounds, providing selectivity scores. The pharmacophore model underwent rigorous testing against active and decoy datasets. The active set comprised 40 active ligands (Supplementary Fig. S1) based on corresponding kinact/KI data29, indicating their potency as PAD2 inhibitors (Supplementary Table 1), while the decoy set encompassed 1000 compounds sourced from Schrodinger’s Decoys Set data59.
The selected model from the previous step indicated reasonable quality with an AUC value of 0.972, and it was indicated to be a great model from the ROC curve analysis (Fig. 2d). Additionally, it successfully identified all the 40 active molecules from the total data set consisted of 1036 compounds (Supplementary Table 2). This validated the reliability of the chosen pharmacophore model (Pharm_01) in predicting active PAD2 compounds.
Virtual screening and drug likeness filter
In the dynamic landscape of drug discovery, virtual screening emerges as a powerful computational technique, effectively navigating vast compound libraries to pinpoint potential drug candidates. However, identifying promising compounds necessitates a thorough evaluation, as not all hits are conducive to further development60. The meticulous assessment of the drug potential of any compound involves applying filters, including Lipinski’s rule of five, Veber’s rule, ADMET criteria, and the TopKat® filter. In our comprehensive study, we conducted virtual screening on the ZINC15 (8,900,485 compounds), Otava (270,020 compounds), DrugBank (12,448 compounds), and Selleckchem kinase library (8,335 compounds) a total of 9,191,288 compounds, employing the selected 3D pharmacophore model (Pharm_01) through DS 4.0’s “Search 3D database” and “Screen Library modules” (Table 2). Integrating the ‘align method parameter’ optimized the alignment of retrieved ligands over the applied pharmacophores, successfully mapping a total of 52,390 compounds from the four databases. Following the methodology outlined, the mapped hits were subjected to stringent filtering to adhere to drug-like properties, resulting in the identification of 2,575 hits. The filtering strategies included Veber’s rule, Lipinski’s rule of five, TopKat® filter, ADMET filter, removing duplicates, and considering a pharmacophoric fit value cutoff of 2.5. Subsequently, molecular docking was applied to these 2,575 hits (Table 2).
This strategic approach facilitates the identification of potential drug candidates and ensures that the selected compounds align with critical drug-like criteria. By subjecting the hits to rigorous filters, we enhance the likelihood of identifying compounds with favorable pharmacokinetic profiles and increased potential for further development61.
Molecular docking using LibDock and CDOCKER
In the intricate drug discovery landscape, molecular docking is a critical predictive tool for understanding the potential interactions between ligands and target macromolecules62. Filtered compounds were docked into the predicted binding site of PAD2, and the grid sphere was included to define the binding site’s configuration (Fig. 3a–c). This study harnessed the capabilities of the LibDock and CDOCKER modules within DS 2020 to unravel these crucial interactions. The LibDock score 82.5513 and CDOCKER score (-CDOCKER energy, 12.634 kcal/mol) of AFM-30a (control) were set as threshold values to find hits with higher affinities.

Visualization of the PAD2 protein structure, showcasing the predicted binding site with grid spheres and key binding site residues. (a) Complete PAD2 protein structure with the grid sphere representing the predicted binding site. (b) Close-up view of the protein binding site with the grid sphere highlighting the docking region. (c) Detailed view of the binding site residues with side chains within the binding pocket.
LibDock, a precision-focused module, efficiently narrowed the initial pool of 2575 hits to 257, concentrating on higher binding affinities. Subsequently, the more refined and accurate CDOCKER tool was employed on these 257 his, contributing to the further refinement of docking structures and mitigating false positives. The computation of empirical scores led to the selection of 92 compounds, each exhibiting higher CDOCKER scores than the control (refer to Supplementary Table 3).
In-depth 2D interaction analyses of the top 92 compounds revealed eight noteworthy candidates with distinct interactions with the Cys-His-Asp catalytic triad (Supplementary Table 4). This critical triad, which consists of His471, Asp473, and Cys647, is essential for the catalytic function of the enzyme. Among these, Lead 1, 2, and 7, previously explored for their anti-cancer properties, were identified from DrugBank and Selleckchem databases63,64,65. Leads 3–6 originated from Otava, while Lead 8 was sourced from ZINC15. The identification of these compounds underscores their potential anti-PAD2 activity, urging further investigation. Moreover, additional significant interactions with the PAD2 protein were unveiled (Fig. 4), further emphasizing their potential efficacy.

2D receptor-ligand interactions of the final hit compounds in the active site of PAD2.
This meticulous exploration enriches our understanding of potential drug candidates and sets the stage for focused investigations into their therapeutic potential against PAD2, advancing our pursuit of novel anti-PAD2 agents.
Covalent docking using flare
In covalent docking, the ligand is modeled to form a covalent bond with a specific amino acid residue in the protein’s active site. Flare 5.066 combines molecular mechanics and quantum chemical calculations to model these interactions. In this study, we targeted the conserved Cys647 residue in the active site of PAD2, crucial for its catalytic activity, as it forms covalent bonds during the citrullination reaction.
Covalent docking was performed using the covalent docking protocol of Flare 5.0 with PAD2 protein and the Cys647 residue as the covalent target. The covalent library from the Selleckchem database was used to generate the ligand structures. A total of 843 prepared covalent ligands were docked into the defined binding site of the PAD2 protein, and the resulting binding poses were ranked based on the lead finder virtual screening (LF-VS) score and binding energy (Dg) score. Among the 843 compounds tested, only two (Lead 9 and Lead 10) listed in Supplementary Table 5 exhibited binding energy (Dg) greater than that of the control ligand; AFM-30a (Dg = -8.48 kcal/mol, and LF-VS = -9.337).
The covalent docking simulations suggest that both Lead 9 and Lead 10 are capable of forming covalent bonds with the Cys647 residue of PAD2. Specifically, the covalent interaction likely involves a nucleophilic attack from the thiol (-SH) group of Cys647 on an electrophilic site on the lead molecules. Given the chemical structures of Lead 9 and Lead 10, it is likely that the covalent bond forms through a mechanism such as Michael’s addition, where the thiol group of Cys647 reacts with an electrophilic carbon atom, resulting in a stable covalent complex. This aligns with the predicted interaction patterns shown in Supplementary Fig. S2, where the covalent bonding interactions are represented in grey. For Lead 9, the reactive group is well-positioned to interact directly with the thiol group of Cys647, forming a covalent bond. This interaction is stabilized by additional hydrogen bonds with residues such as Asp351, Arg373, Asn590, and Leu642, alongside hydrophobic interactions with Trp348, His471, Phe641, Asp473, Glu474, Val469, Thr403, Arg441, and Asp578. Similarly, Lead 10 is also predicted to form a covalent bond with Cys647, with further stabilization provided by hydrogen bonds involving residues Phe573, Arg582, Glu644, Asn587, Asn590, Asp473, Phe641, His646, Ser407, Thr403, and Asp578.
The docking results suggest a high likelihood that Lead 9 and Lead 10 can form stable covalent complexes with PAD2, supported by favorable binding modes, hydrogen bonding, hydrophobic interactions, and electrostatic interactions, as detailed in Supplementary Tables 4 and visualized in Fig. 4.
MD Simulation studies
Following molecular docking studies, we investigated the stability and dynamics of protein-ligand complexes through Molecular Dynamics (MD) simulations. This analysis included the top 8 compounds from pharmacophore-based screening and 2 from covalent docking, and each simulated for 100 ns. Comparative analyses were conducted against PAD2 bound to the control ligand (AFM-30a). The evaluation criteria encompassed the Energy Profile, RMSD, RMSF, Radius of Gyration (Rg), and interactions involving protein-protein/protein-ligand hydrogen bonds.
The energy profile graph depicts the potential energy of the receptor-ligand complexes and the control compound (AFM-30a) over the 100 ns simulation time. The potential energy represents the interaction energy between the protein and ligand molecules. The potential energy of receptor-ligand complexes, including the control (AFM-30a), remained stable (~ -9.55e + 05 kJ/mol) post an initial equilibration period, indicating overall simulation stability (Supplementary Fig. S3).
The RMSD values represent the average distance between the receptor-ligand complex structures at various simulation time points67. The RMSD values were computed about the initial structure before the simulation. RMSD analysis revealed stable receptor-ligand complexes over 100 ns, with no significant structural changes. Seven of the compounds (Lead 2, Lead 3, Lead 5, Lead 6, and Lead 8–10) complexed with PAD2 were more stable as they had lower or similar average RMSD values in comparison to PAD2-control complex. The average RMSD values for the other three compounds (Lead 1, Lead 4, and Lead 7) were relatively higher, indicating less stability than the control compound (Fig. 5). All the complexes were very well within limits with RMSD below 0.6.

RMSD analysis of the filtered compounds throughout the 100ns MD run.
In RMSF analysis, the flexibility and stability of a protein-ligand complex are evaluated by looking at how individual protein residues change over time. Residue fluctuation is the term used to describe changes in each amino acid residue’s position over time. While low RMSF values signify that the residues are more rigid and stable, high RMSF values indicate that some residues are more flexible and exhibit greater fluctuations68. In this case, we have evaluated the RMSF plot for the top 10 protein-ligand complexes along with control compounds that were simulated for 100 ns each (Supplementary Fig. 4). The fluctuating residues from the RMSF plot for the ten complexes and the control complex are shown in Supplementary Table 6.
The table shows that some residues like Lys322, Glu323, and Pro665 frequently fluctuate across different complexes. Still, none of the fluctuating residues were present in the binding pocket, supporting that all the complexes were remarkably stable during the 100 ns MD run.
The spatial extent of a protein or protein-ligand complex is measured by the Radius of Gyration (Rg). It gives more knowledge about the overall size, shape, flexibility, and compactness of the complex69. Rg measurements indicated that all complexes were highly compact and rigid, with PAD2-Lead 3, PAD2-Lead 4, and PAD2-Lead 5 showing Rg values like the PAD2-control (AFM-30a). Other complexes exhibited higher average Rg values, implying varying degrees of compactness and flexibility (Supplementary Fig. S5).
In MD simulations, protein-protein hydrogen bonds can reveal important details about the dynamics and stability of protein complexes. Based on the hydrogen bond analysis performed throughout the 100 ns run (Supplementary Fig. 6), it was found that all the complexes displayed a consistent pattern (~ 250) of potent intermolecular hydrogen bonds that persisted over time. This demonstrates that the protein-protein interactions were robust and preserved the complex’s structural integrity.
Our results (Fig. 6) showed that the number of protein-ligand hydrogen bonds varied significantly among the ten complexes studied. The top three complexes with the highest number of hydrogen bonds formed were PAD2-Lead 2, PAD2-Lead 5, and PAD2-Lead 8. On the other hand, PAD2-Lead 4 and PAD2-Lead 10 had a lower number of H-bonds than the PAD2-control complex. In the case of top lead compounds (Lead 2, Lead 5, and Lead 8), four to five H-bonds were stable throughout the simulation compared to the control compound (two to three H-bonds). The results suggest that the stability of a protein-ligand complex is significantly influenced by the number of hydrogen bonds formed between a protein and its ligand.

Protein-protein H-bond analysis of the filtered compounds throughout the 100ns MD run.
Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA)
The binding free energy between the ligand and receptor is inferred from the ∆Gbind-PBSA values calculated from the MM-PBSA simulations. A more negative ∆Gbind-PBSA value indicates a stronger binding affinity.
The results illustrated in Table 3 indicate that the ∆Gbind-PBSA value of the ten ligand-receptor complexes ranges from highly negative (-297.484 kJ/mol for Lead 2 and − 234.347 kJ/mol for Lead 8) to comparatively less negative (-79.244 kJ/mol for Lead 7). The control-protein complex has a moderately negative Gbind-PBSA value (-117.775 kJ/mol), indicating its comparatively lower affinity. Lead 1 (Olomoucine II) and Lead 2 (Capivasertib) from the DrugBank database and Lead 9 (THZ1) from the Selleckchem database have already been investigated for anticancer properties. Therefore, these hit molecules can be repurposed as PAD2 inhibitors. These findings imply that the strength of the ligand-receptor interactions in the ten complexes varies, with some having strong binding affinities and others having weaker binding affinities.
Additionally, the MM-PBSA method was employed to analyze the energy contributions of individual residues within the binding site. We focused on the top 4 hit compounds (Lead 1, Lead 2, Lead 8, and Lead 9) for this analysis. The per-residue energy decomposition was performed to assess the interactions in which each residue is involved. Residues exhibiting significant energy deviations are highlighted in Supplementary Figure S7, where green indicates a negative energy contribution (favorable for binding) and red indicates a positive energy contribution (unfavorable for binding). Key residues contributing to favorable interactions across the lead compounds were identified, including Asp346, Asp351, Glu352, Asp465, Asp473, Glu474, Asp576, Glu577, Asp578, and Met521.
Previous studies on PAD2 inhibitors have identified several key residues involved in the enzyme’s activity. Residues such as Asp351, Asp473, Met521, and Cys647 are crucial for PAD2 function and inhibitor binding. Our analysis also shows that residues like Asp351, Asp473, and Met521 contribute significantly to the binding of the lead compounds, which aligns with their known roles in stabilizing ligand interactions. This consistency highlights the importance of these conserved residues in PAD2 inhibition and supports the relevance of our computational findings.
Lead 2 and 9 demonstrated the maximum favorable interactions, largely due to their high electrostatic energy contributions, significantly enhancing their binding affinity. Lead 1 and 8 displayed a similar pattern in the graph, indicating comparable binding interactions. This detailed energy decomposition provides deeper insights into the key residues driving the binding affinities of the top-hit compounds, offering valuable information for further optimization and development of PAD2 inhibitors.
Principal component analysis
The 2D projection of the Principal Component Analysis (PCA) plot from the molecular dynamics (MD) simulation visualizes the trajectory of the selected top five leads (Lead 1, Lead 2, Lead 5, Lead 8, and Lead 9) and the control compound (AFM-30a). This selection is based on MM-PBSA analyses and represents their projections onto two principal components, eigenvector 1 and eigenvector 2 (Fig. 7).

PCA plot illustrating the most significant principal components of motion for the Cα atoms of PAD2 in complex with the control compound (AFM-30a) and selected lead compounds.
The control compound, represented by black points, is spread around the origin, indicating central and dispersed data around the zero mark on both eigenvector axes. Lead 1 (Olomoucine II), depicted as yellow points, divides into many clusters with one center dense area and scattered points, showing a substantial divergence from the control and higher variance along the eigenvector 2 axis. Lead 2 (Capivasertib), represented by red points, shows few intermixed points, suggesting minimal deviation from the control and other leads. Lead 8, represented by green points, exhibits distinct clusters, with one close to the center and another spread out to the right, indicating a unique trajectory compared to other leads and the control. Lead 9 (THZ1), illustrated by blue points, produces a tight cluster cantered on the eigenvector 1 axis with a minor spread, indicating some fluctuation but generally remaining close to the control group. Lead 5, depicted by brown points, creates a compact cluster near the center with dispersed points around it, indicating a deviation from the control similar to Lead 1 but less spread out.
The PCA plot reveals how each lead deviates from the control. The distinct clustering of green points (Lead 8) suggests that this lead has a unique response in the simulation. In contrast, other leads show varying degrees of overlap (Lead 2 and Lead 5) and dispersion (Lead 1 and Lead 9) around the control group, indicating that their responses are more similar to the control or less distinct from each other. The spread and clustering of points provide insights into the variability and similarity of molecular dynamics under different conditions. Closer points indicate more similar behaviours, while distinct clusters or spread-out points indicate significant deviations and unique behaviours.
The 3D free energy landscapes (Fig. 8) for the control compound (AFM-30a) and selected lead compounds (Lead 1, Lead 2, Lead 5, Lead 8, and Lead 9) reveal differences in their energetic profiles when projected onto the principal components (PC1 and PC2). The control compound exhibits a relatively centralized energy landscape with a predominant low-energy basin, indicating stability around the origin. Lead 1 and 8 have a more confined and deeper energy basin, indicating a stable state with less conformational variation. Lead 5 presents a landscape similar to the control but with slightly broader energy minima, indicating stability with minor variations. Lead 2 and 9 exhibit a spread and varied landscape with multiple energy basins, suggesting higher conformational diversity. Among all the leads, Lead 1 and 8 stand out with significant and deep energy minima extending along both PC1 and PC2, indicating a unique and highly stable conformational behaviour. This implies that Lead 1 and 8 potentially have the most significant effect and could be considered the best candidates for further development based on their distinctive and stable free energy profile compared to the control and other leads.

Gibbs free energy landscape. (a) Control, (b) Lead 1, (c) Lead 2, (d) Lead 5, (e) Lead 8, and (f) Lead 9.
Binding mode analyses
The binding mode analyses provided important insights into the binding interactions of the lead compounds (Lead 1, Lead 2, Lead 8, and Lead 9) with Peptidyl Arginine Deiminase 2 (PAD2).
The benzimidazole group in the control compound AFM-30a (MMPBSA binding affinity − 111.775 kcal/mol) was positioned towards Thr468 and Arg582, resulting in van der Waals interactions. The fluoroacetamidine group formed a hydrophobic network with Cys647, Val469, and 469, as well as a hydrogen bond with Asp351 and Leu642. This group also directed towards important His471 leading to van der Waals interaction. The isoindolinone ring system consists of a benzene ring, which formed a carbon-hydrogen bond with Trp348 and a Pi-Pi T-shaped bond with Phe641. Phe641 also interacted via carbon-hydrogen with the free alkyl group in the imidazole ring.
Lead 2 demonstrated the highest binding affinity of -297.484 kcal/mol in the MMPBSA analysis. Its purine ring, which is made up of a pyrimidine ring fused to an imidazole ring, formed most of the crucial interactions with PAD2. Specifically, these two rings made strong Pi-alkyl bonds with Val469. The pyrimidine ring also formed key Pi-Pi stacking interactions with Trp348 and Phe641, a significant Pi-sulfur interaction with Cys647, a conventional hydrogen bond with Asp351, and a carbon-hydrogen bond with Leu642. All these interactions played a vital role in PAD2 selectivity. Additionally, the pyrimidine ring of Lead 2 exhibited important carbon-hydrogen bonds with Ser407 and His471, which are critical for its inhibitory activity. The presence of conventional hydrogen bonds with Lys640 and Phe641 further increased binding affinity compared to AFM-30a29. Like AFM-30a, Lead 2, through its chlorophenyl group, contributed to electrostatic interactions with the active site residue Arg347, further stabilizing the inhibitor-enzyme complex. Moreover, van der Waals interactions with critical residues Asp473 and Arg373 further enhanced the binding of Lead 2 to PAD2. This molecule demonstrated better interactions within the binding site as compared to the positive control AFM-30a, suggesting that Lead 2 could serve as a potent and reversible inhibitor of PAD2.
Lead 8 with the next best binding affinity (-234.347 kcal/mol-1) determined by the MMPBSA method. This affinity was driven by its ability to form key hydrogen bonds with Trp348 (carbon-hydrogen bond) and Phe641 (conventional hydrogen bond), facilitated by its benzene ring and amide group, respectively. Lead 8 also exhibited strong hydrophobic interactions with Val469, Cys647, and Leu642, with the help of its methoxy group and aromatic ring. The terminal aromatic ring of Lead 8 played a crucial role by forming important Pi-alkyl interactions with Trp348 and alkyl bonds with Phe641, both of which are essential for PAD2 selectivity. Additionally, the isopropyl group attached to one of the benzene rings oriented itself towards significant residues like His471, Arg373, Asp351, Ser404, and Ser407, resulting in van der Waals interactions.
Lead 9 showed a binding affinity of -187.703 kcal/mol calculated using the MMPBSA method. Its benzimidazole ring played a pivotal role by forming dual Pi-Pi stacking interactions with Trp348 and a Pi-sulfur bond with Cys647 and Val469, which are crucial for PAD2 selectivity. Through van der Waals interactions, this group also aligned itself towards critical binding site residues like Asp351, His471, and Asp473. Additionally, Lead 9 formed strong hydrogen bonds and Pi-Pi stacking interactions with Phe641. The aromatic ring, featuring a charged chlorine atom, established an important Pi-cation bond with Arg373. Furthermore, the amide group in Lead 9 exhibited dual strong hydrogen bonds with Lys640.
Lead 1, with a reasonable binding affinity of -143.891 kcal/mol as calculated by MMPBSA analysis, exhibited crucial interactions with key PAD2 residues important for selectivity. The hydroxyl group attached to its aromatic ring formed a strong hydrogen bond with Asp346. Notably, Trp348, a critical residue for PAD2 inhibition, formed a solid bond, dual carbon-hydrogen bonds with the amide group and aromatic ring, and a Pi-alkyl bond with the terminal alkyl chain. The alkyl chain also engaged in hydrophobic interactions with His471 and Val469. Additionally, the hydroxyl group on the alkyl chain established dual carbon-hydrogen bonds with Asp351 and a conventional hydrogen bond with Arg373, both of which are vital for high binding affinity against PAD2. Other important van der Waals interactions involved residues such as Cys647, Asp473, Phe641, Asn376, and Ser407, contributing to Lead 1’s selectivity for PAD2. These interactions collectively underscore Lead 1’s potential as a selective PAD2 inhibitor.
To summarise, the binding mode analyses, Fig. 9 emphasizes the importance of hydrogen bonding, hydrophobic interactions, and van der Waals interactions in the lead compounds binding affinities to PAD2. These interactions involve critical residues such as Gly345, Asp346, Arg347, Trp348, Asp351, Arg373, Ser404, Ser407, Thr468, Val469, His471, Asp473, Phe641, Leu642, and Cys647, which are required for effective PAD2 inhibition. Comparing the lead compounds to the control molecule AFM-30a reveals similar binding interactions, emphasizing the role of aromatic systems, halogen bonds, and polar interactions in improving binding stability and specificity. Furthermore, Lead 2 and 9 emerged as better potential inhibitors than AFM-30a due to their additional hydrogen bonds with Lys640, which significantly increase the stability of the inhibitor-enzyme complex. Lead 2 and 9 also possess Pi-sulfur and Pi-cation interactions that are absent in AFM-30a, enhancing their binding affinity and selectivity for PAD2. Consistent with these findings, the MM-PBSA results (Supplementary Figure S7) also showed that Lead 2 and 9 demonstrated the maximum favorable interactions, largely due to their high electrostatic energy contributions, significantly enhancing their binding affinity. These interactions underscore the potential of Lead 2 and 9 as potential and selective PAD2 inhibitors. This extensive study of the lead compounds’ binding interactions and structural characteristics lays the groundwork for future optimization and development of selective PAD2 inhibitors.

Binding mode analyses of lead 2 compared to other leads and control compound (AFM-30a). The color-coded interactions underscore the significant contributions of hydrogen bonds (green color), pi-alkyl (pink color), pi-pi stacking bond (dark pink color), pi-sulfur (orange color), and pi-cation (golden color) in the inhibitory activity of these lead compounds against PAD2.
Conclusion
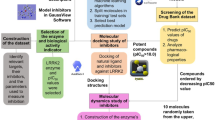
This study presents an in-depth in silico exploration targeting the peptidyl arginine deiminase 2 (PAD2) enzyme, which plays a crucial role in various diseases. We developed a structure-based pharmacophore model, as illustrated in Fig. 10, which guided the identification of potential PAD2 inhibitors through database screening. Covalent docking and molecular dynamics simulations further refined our selection, highlighting several lead compounds with strong binding potential and stability.

Graphical summary of the study on PAD2 inhibitor discovery. This figure highlights the stepwise approach, including pharmacophore-based virtual screening, molecular docking, molecular dynamics simulations, and binding energy calculations, leading to the identification of PAD2 inhibitors.
Notably, we identified two hit compounds (Lead 1 and 2) from the DrugBank database that show promise for drug repurposing, and a potential novel inhibitor (Lead 8) from the ZINC database. However, it is important to emphasize that these results suggest potential inhibition and further experimental validation is needed to confirm the inhibitory potential of these compounds against PAD2. Overall, this research provides important insights into PAD2 inhibition and lays a foundation for future experimental studies to develop new therapeutic options.
Data availability
All data analyzed or generated during this study are included in this article and its supplementary file.
References
Rogers, G. E., Harding, H. W. J. & Llewellyn-Smith, I. J. The origin of citrulline-containing proteins in the hair follicle and the chemical nature of trichohyalin, an intracellular precursor. Biochim. Biophys. Acta BBA Protein Struct. 495, 159–175 (1977).
Rogers, G. E. & Simmonds, D. H. Content of citrulline and other amino-acids in a protein of hair follicles. Nature 182, 186–187 (1958).
Mansouri, P. et al. Peptidylarginine deiminase (PAD): A promising target for chronic diseases treatment. Int. J. Biol. Macromol. 278, 134576 (2024).
Olsen, I., Singhrao, S. K. & Potempa, J. Citrullination as a plausible link to periodontitis, rheumatoid arthritis, atherosclerosis and Alzheimer’s disease. J. Oral Microbiol. 10, 1487742 (2018).
Yuzhalin, A. E. Citrullination in cancer. Cancer Res. 79, 1274–1284 (2019).
Rus’d, A. A. et al. Molecular cloning of cDNAs of mouse peptidylarginine deiminase type I, type III and type IV, and the expression pattern of type I in mouse. Eur. J. Biochem. 259, 660–669 (1999).
Ishigami, A. et al. Human peptidylarginine deiminase type II: Molecular cloning, gene organization, and expression in human skin. Arch. Biochem. Biophys. 407, 25–31 (2002).
Chavanas, S. et al. Comparative analysis of the mouse and human peptidylarginine deiminase gene clusters reveals highly conserved non-coding segments and a new human gene, PADI6. Gene 330, 19–27 (2004).
Gijon, M., Metheringham, R. L., Toss, M. S., Paston, S. J. & Durrant, L. G. The clinical and prognostic significance of protein arginine deiminases 2 and 4 in colorectal cancer. Pathobiology 89, 38–48 (2022).
Teng, Y., Chen, Y., Tang, X., Wang, S. & Yin, K. PAD2: A potential target for tumor therapy. Biochim. Biophys. Acta Rev. Cancer 1878, 188931 (2023).
Slade, D. J. et al. Protein arginine deiminase 2 binds calcium in an ordered fashion: Implications for inhibitor design. ACS Chem. Biol. 10, 1043–1053 (2015).
Ying, S. et al. Transcriptional regulation of peptidylarginine deiminase expression in human keratinocytes. J. Dermatol. Sci. 53, 2–9 (2009).
Zhang, Y. et al. PADs in cancer: Current and future. Biochim. Biophys. Acta Rev. Cancer 1875, 188492 (2021).
Cherrington, B. D. et al. Potential role for PAD2 in gene regulation in breast cancer cells. PLoS ONE 7, (2012).
Watanabe, K. et al. Combined biochemical and immunochemical comparison of peptidylarginine deiminases present in various tissues. Biochim. Biophys. Acta 966, 375–383 (1988).
Zhang, X. et al. Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor α target gene activation. Proc. Natl. Acad. Sci. 109, 13331–13336 (2012).
Khajavi, M. et al. Identification of Padi2 as a novel angiogenesis-regulating gene by genome association studies in mice. PLoS Genet. 13, e1006848 (2017).
McElwee, J. L. et al. Identification of PADI2 as a potential breast cancer biomarker and therapeutic target. BMC Cancer 12, 500 (2012).
Wang, H. et al. PADI2 gene confers susceptibility to breast cancer and plays tumorigenic role via ACSL4, BINC3 and CA9 signaling. Cancer Cell. Int. 16, 61 (2016).
Horibata, S. et al. Role of peptidylarginine deiminase 2 (PAD2) in mammary carcinoma cell migration. BMC Cancer 17, 378 (2017).
Li, F. et al. Inhibiting PAD2 enhances the anti-tumor effect of docetaxel in tamoxifen-resistant breast cancer cells. J. Exp. Clin. Cancer Res. 38, 414 (2019).
Wang, L. et al. PADI2-Mediated citrullination promotes prostate cancer progression. Cancer Res. 77, 5755–5768 (2017).
Urick, M. E. & Bell, D. W. Proteomic profiling of FBXW7-mutant serous endometrial cancer cells reveals upregulation of PADI2, a potential therapeutic target. Cancer Med. 9, 3863–3874 (2020).
Asad, M., Wajid, S., Katare, D. P., Mani, R. J. & Jain, S. K. Differential expression of TOM34, AL1A1, PADI2 and KLRBA in NNK induced lung cancer in wistar rats and their implications. Curr. Cancer Drug Targets 19, 919–929 (2019).
Mondal, S. & Thompson, P. R. Protein arginine deiminases (PADs): Biochemistry and chemical biology of protein citrullination. Acc. Chem. Res. 52, 818–832 (2019).
Jang, B., Ishigami, A., Kim, Y. S. & Choi, E. K. The peptidylarginine deiminase inhibitor Cl-Amidine suppresses inducible nitric oxide synthase expression in dendritic cells. Int. J. Mol. Sci. 18, 2258 (2017).
Martín Monreal, M. T. et al. Applicability of small-molecule inhibitors in the study of peptidyl arginine deiminase 2 (PAD2) and PAD4. Front. Immunol. 12, (2021).
Witalison, E. E., Thompson, P. R. & Hofseth, L. J. Protein arginine deiminases and associated citrullination: Physiological functions and diseases associated with dysregulation. Curr. Drug Targets 16, 700–710 (2015).
Muth, A. et al. Development of a selective inhibitor of protein arginine deiminase 2. J. Med. Chem. 60, 3198–3211 (2017).
Teo, C. Y. et al. Discovery of a new class of inhibitors for the protein arginine deiminase type 4 (PAD4) by structure-based virtual screening. BMC Bioinform. 13(Suppl 17), S4 (2012).
Rahman, M. B. A. C. Y. T. Ligand-based virtual screening for the discovery of inhibitors for protein arginine deiminase type 4 (PAD4). in J. Postgenomics Drug Biomark. Dev. 03 (2013).
Systèmes, D. BIOVIA, discovery studio visualizer, release 2019. San Diego Dassault Systèmes (2020).
Van Der Spoel, D. et al. GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005).
Pal, S. et al. Ligand-based pharmacophore modeling, virtual screening and molecular docking studies for discovery of potential topoisomerase I inhibitors. Comput. Struct. Biotechnol. J. 17, 291–310 (2019).
Arooj, M., Sakkiah, S., Kim, S., Arulalapperumal, V. & Lee, K. W. A combination of receptor-based pharmacophore modeling & QM techniques for identification of human chymase inhibitors. PLoS ONE 8, e63030 (2013).
Bouback, T. A. et al. Pharmacophore-based virtual screening, quantum mechanics calculations, and molecular dynamics simulation approaches identified potential natural antiviral drug candidates against MERS-CoV S1-NTD. Molecules 26, 4961 (2021).
Tai, W. et al. Pharmacophore modeling and virtual screening studies to identify new c-Met inhibitors. J. Mol. Model. 18, 3087–3100 (2012).
Sterling, T. & Irwin, J. J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Volochnyuk, D. M. et al. Evolution of commercially available compounds for HTS. Drug Discov Today 24, 390–402 (2019).
Seidel, T., Ibis, G., Bendix, F. & Wolber, G. Strategies for 3D pharmacophore-based virtual screening. Drug Discov Today Technol. 7, e221–e228 (2010).
Hiremath, C. N. Abbreviated Profile of drugs (APOD): modeling drug safety profiles to prioritize investigational COVID-19 treatments. Heliyon 7, e07666 (2021).
Jha, P., Saluja, D. & Chopra, M. Structure-guided pharmacophore based virtual screening, docking, and molecular dynamics to discover repurposed drugs as novel inhibitors against endoribonuclease Nsp15 of SARS-CoV-2. J. Biomol. Struct. Dyn. 0, 1–11 (2022).
Rao, S. N., Head, M. S., Kulkarni, A. & LaLonde, J. M. Validation studies of the site-directed docking program LibDock. J. Chem. Inf. Model. 47, 2159–2171 (2007).
Wu, G., Robertson, D. H., Brooks, I. I. I., Vieth, M. & C. L. & Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 24, 1549–1562 (2003).
Alamri, M. A. et al. Discovery of anti-MERS-CoV small covalent inhibitors through pharmacophore modeling, covalent docking and molecular dynamics simulation. J. Mol. Liq. 330, 115699 (2021).
Hamdi, A. et al. Design, synthesis, antitumor, and VEGFR-2 inhibition activities of novel 4-anilino-2-vinyl-quinazolines: Molecular modeling studies. Bioorg. Chem. 122, 105710 (2022).
Software. https://www.cresset-group.com/software/
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015).
Schuler, L. D., Daura, X. & van Gunsteren, W. F. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 22, 1205–1218 (2001).
Fuhrmans, M., Sanders, B. P., Marrink, S. J. & de Vries, A. H. Effects of bundling on the properties of the SPC water model. Theor. Chem. Acc. 125, 335–344 (2010).
Simmonett, A. C. & Brooks, B. R. A compression strategy for particle mesh Ewald theory. J. Chem. Phys. 154, 054112 (2021).
Genheden, S. & Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 (2015).
David, C. C. & Jacobs, D. J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. Clifton NJ 1084, 193–226 (2014).
Shafiee, Z. et al. Insights into the molecular mechanism of triazolopyrimidinone derivatives effects on the modulation of α1β2γ2 subtype of GABAA receptor: An in silico approach. Arch. Biochem. Biophys. 729, 109380 (2022).
Konc, J. Binding site comparisons for target-centered drug discovery. Expert Opin. Drug Discov. 14, 445–454 (2019).
Baby, S. T., Sharma, S., Enaganti, S. & Cherian, P. R. Molecular docking and pharmacophore studies of heterocyclic compounds as Heat shock protein 90 (Hsp90) inhibitors. Bioinformation 12, 149–155 (2016).
Chopra, M., Gupta, R., Gupta, S. & Saluja, D. Molecular modeling study on chemically diverse series of cyclooxygenase-2 selective inhibitors: generation of predictive pharmacophore model using Catalyst. J. Mol. Model. 14, 1087–1099 (2008).
Réau, M., Langenfeld, F., Zagury, J. F., Lagarde, N. & Montes, M. Decoys selection in benchmarking datasets: Overview and perspectives. Front. Pharmacol. 9, (2018).
Hevener, K. E. et al. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model. 49, 444–460 (2009).
Rester, U. From virtuality to reality - virtual screening in lead discovery and lead optimization: A medicinal chemistry perspective. Curr. Opin. Drug Discov. Devel. 11, 559–568 (2008).
Muegge, I. Selection criteria for drug-like compounds. Med. Res. Rev. 23, 302–321 (2003).
Meng, X. Y., Zhang, H. X., Mezei, M. & Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 7, 146–157 (2011).
Macip, G. et al. Haste makes waste: a critical review of docking-based virtual screening in drug repurposing for SARS-CoV-2 main protease (M-pro) inhibition. Med. Res. Rev. 42, 744–769 (2022).
Andrikopoulou, A. et al. The emerging role of capivasertib in breast cancer. Breast Off. J. Eur. Soc. Mastology. 63,157–167 (2022).
Blunt, M. D. et al. The Dual Syk/JAK Inhibitor Cerdulatinib Antagonizes B-cell Receptor and MicroenvironmentalSignaling in Chronic Lymphocytic Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 23, 2313–2324 (2017).
Holcakova, J. et al. The inhibitor of cyclin-dependent kinases, olomoucine II, exhibits potent antiviral properties.Antivir. Chem. Chemother. 20, 133–142 (2010).
Sargsyan, K., Grauffel, C. & Lim, C. How molecular size impacts RMSD applications in molecular dynamics simulations. J. Chem. Theory Comput. 13, 1518–1524 (2017).
Martínez, L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE 10, e0119264 (2015).
van Gunsteren, W. F., Dolenc, J. & Mark, A. E. Molecular simulation as an aid to experimentalists. Curr. Opin. Struct. Biol. 18, 149–153 (2008).
Acknowledgements
Authors thankfully acknowledge the Bioinformatics Centre (BIC), National Network Project (NNP), Department of Biotechnology, Govt. of India, and Institution of Eminence (IoE), University of Delhi to MC for financial support. The authors acknowledge Dr. B. R. Ambedkar Center for Biomedical Research, University of Delhi, for lending bioinformatics infrastructural facility (BIC) and Bioinformatics Resources and Applications Facility (BRAF), C-DAC, ‘Pune’ for providing server assistance. PJ received a PhD fellowship from DST-Inspire by grant number (DST/INSPIRE/03/2016/000026). PR wants to acknowledge the Department of Biotechnology (DBT), Ministry of Science & Technology, Government of India for the DBT-JRF fellowship (Grant Number: DBT/2021–22/CBR/1593).
Funding
This work was supported by a Grant from the Department of Biotechnology (Govt. of India) for the Establishment of Bioinformatics Centre (BIC) (BT/PR40153/BTIS/137/8/2021), National Network Project (BT/PR40195/BTIS/137/58/2023) at Dr. B. R. Ambedkar Center for Biomedical Research and the Institute of Eminence, University of Delhi [IoE/2023-24/12/FRP].
Author information
Authors and Affiliations
Contributions
P.J. and M.C. conceived and designed the experiments. M.C. contributed towards supervision. P.J. conducted all the experiments and was responsible for writing the entire manuscript and preparing the figures and tables. P.J., P.R., P.P., and M.C., all contributed to the writing, review, and editing process.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jha, P., Rajoria, P., Poonia, P. et al. Identification of novel PAD2 inhibitors using pharmacophore-based virtual screening, molecular docking, and MD simulation studies. Sci Rep 14, 28097 (2024). https://doi.org/10.1038/s41598-024-78330-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-78330-5
