
Abstract
Previous studies have suggested a potential association between the gut microbiota and arthritis. However, the causal links between the gut microbiota and various types of arthritis, as well as the potential mediating role of inflammatory proteins, remain unclear. Mendelian randomization was used to explore the causal relationships between gut microbiota, inflammatory proteins, and various forms of arthritis (osteoarthritis, rheumatoid and psoriatic arthritis, and ankylosing spondylitis [AS]). The inverse variance-weighted method was the primary analytical approach used. Furthermore, we examined the mediating role of inflammatory proteins in the pathway linking the gut microbiota to arthritis. Sensitivity analyses were performed to verify the robustness of the findings, and enrichment analyses were conducted to investigate the biological functions and pathways involved. We identified 11 positive and 14 negative causal effects linking the genetic liability of the gut microbiota to arthritis. Similarly, 9 positive and 8 negative causal effects between inflammatory proteins and arthritis were identified. Notably, an increased abundance of the order Bacillales (odds ratio [OR] = 1.199, 95% confidence interval [CI] = 1.030–1.394, P = 0.019) and higher interleukin-7 levels (OR = 1.322, 95% CI = 1.004–1.741, P = 0.046) significantly elevated the risk of AS. Furthermore, interleukin-7 mediated 13.8% of the effect caused by the order Bacillales, with a mediation effect size of β = 0.025 (95% CI = 0.001–0.064). Sensitivity and supplementary analyses revealed no significant evidence of horizontal pleiotropy or heterogeneity. Overall, our findings demonstrate causal links between the gut microbiota, inflammatory proteins, and four arthritis types, highlighting the gut microbiota as a potential therapeutic target. Crucially, interleukin-7 not only strongly correlated with AS but also partially mediated the effect exerted by the gut microbiota on AS, suggesting that managing the gut microbiota to modulate inflammatory proteins could serve as an effective therapeutic strategy for arthritis.
Similar content being viewed by others


Introduction
In recent decades, rapid advancements in biotechnology and genomics have significantly expanded our understanding of the human microbiome, particularly in the study of the gut flora1. The gut microbiome is one of the largest microbial communities within the human body. It is closely connected to the host immune system and plays a key role in regulating immune balance and maintaining tissue homeostasis2. Numerous studies have demonstrated a close link between the gut microbiome and human health. These microbes are not only involved in digestion, absorption, and metabolic regulation, but are also linked to obesity, diabetes, cardiovascular and inflammatory diseases, and the development of certain types of cancer3.
Recently, inflammatory diseases have become a focal point of research. Among these, arthritis, a common chronic inflammatory disease, has a particularly complex pathogenesis involving interactions between various immune cells and inflammatory mediators4. Recent studies have indicated that imbalances in the gut microbiome play a significant role in the development of arthritis. It indirectly affects disease progression by regulating host immune responses and inflammation levels. Specifically, a series of studies has shown that the gut microbiome composition of patients with rheumatoid arthritis (RA) and ankylosing spondylitis (AS) exhibits significant differences compared to that of healthy individuals5,6. Furthermore, experiments using animal models have revealed that transferring the gut microbiota can significantly alter the condition of arthritis in animals, further confirming the crucial role of the gut microbiome in the development of arthritis4.
However, despite these findings revealing a correlation between the gut microbiome and arthritis, the current research findings have limitations, particularly in proving causality. Most studies are based on observational designs, meaning that they cannot eliminate the interference of confounding factors nor determine directionality, that is, whether imbalances of the gut microbiome lead to arthritis or if the state of arthritis changes the composition of the gut microbiome. The Mendelian randomization (MR) method, which uses genetic variations associated with characteristics of the gut flora as instrumental variables (IVs), offers a new approach for addressing this challenge7,8. The MR method overcomes some of the limitations of traditional observational studies, such as confounding bias and reverse causation issues, thus providing more reliable evidence for a potential causal relationship between the gut microbiome and arthritis.
Notably, an imbalance in the gut microbiome may lead to the release of a series of inflammatory proteins, including but not limited to various interleukin (IL) and tumor necrosis factor (TNF) proteins9. These inflammatory proteins play crucial roles in the pathogenesis of arthritis, affecting its development and progression10. Therefore, an imbalance in the gut microbiome through regulation of the release of inflammatory proteins could be a key link in the mechanism involved in arthritis development. Focusing on inflammatory proteins could not only deepen our understanding of the mechanism of arthritis development but could also provide a new perspective on the causal relationship between gut microbiome imbalance and arthritis.
This study aimed to explore the causal connection between the gut microbiota and arthritis (including osteoarthritis [OA], RA, psoriatic arthritis [PsA], and AS) using the MR method. We further analyzed the mediating role of inflammatory proteins in this process, with the objective of providing new insights into the pathological mechanisms of arthritis and the potential application of the gut microbiota in arthritis treatment.
Methods
Study design
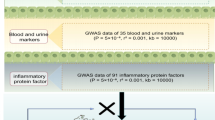
This study was divided into four core sections, as shown in Fig. 1: first, the causal effects of 211 types of gut microbiota on the four types of arthritis were analyzed (Phase 1 A); second, the impact of 91 inflammatory proteins on these arthritis types was explored (Phase 2 A); third, the connection between the gut microbiota and arthritis was investigated, particularly regarding the mediating role of inflammatory markers (Phase 3); finally, genes near the key single nucleotide polymorphisms (SNPs) discovered in Phase 1 A were subjected to gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses (Phase 4). SNPs were defined as IVs. This study was based on three fundamental assumptions of MR: first, the IV must be strongly associated with the exposure factor under study; second, the IV must not be related to any confounding factors; and third, the IVs cannot act directly on the outcome but rather be mediated through specific exposure pathways11.

Flowchart overviewing the Mendelian randomization analysis.
Data sources
The latest genome-wide association study (GWAS) summary data provide a genetic backdrop for the gut microbiome. These data were collected and analyzed by the MiBioGen consortium, which comprises whole-genome genotype information and stool microbiome data based on the 16 S rRNA from 18,340 individuals spread across 24 cohorts12. The compiled data includes 211 different gut microbial taxa categorized into 131 genera, 35 families, 20 orders, 16 classes, and 9 phyla. The genetic information on inflammatory proteins, however, was based on previous GWAS research conducted by Zhao and his team13, covering 91 types of inflammatory proteins. GWAS data for RA, PsA, and AS were sourced from 10th edition resources of the FinnGen consortium (https://r10.risteys.finngen.fi/). The study results on OA (of the hip or knee), however, originated from the research conducted by Tachmazidou I and their team14, which had been incorporated into the IEU Open GWAS database (https://gwas.mrcieu.ac.uk/).
This prospective cohort study screened four types of arthritis using the International Classification of Diseases diagnostic codes and involved a reanalysis of publicly available GWAS summary data. All original GWAS studies involved herein had obtained the necessary ethical consent, and given that this study did not use individual-level data, obtaining further approval from the institutional ethics review board was not necessary.
Genetic instrument selection
In this MR study, the selection of genetic instruments (IVs) was based on the three fundamental assumptions mentioned in the study design. Initially, SNPs that were significantly associated with the gut microbiota (P < 1 × 10–5) were selected. To maximize the number of instruments available for each inflammatory protein, we set the P-value threshold for SNP selection at 5 × 10–6. SNPs exhibiting linkage disequilibrium (LD) were excluded from the analysis. When selecting SNPs closely related to the gut microbiota and inflammatory proteins, we ensured that their LD met the following criteria: r2< 0.001 and a distance greater than 10,000 kb. An important step in the MR analysis was to ensure that the effect of the SNPs on exposure corresponded to the same allele as their effect on the outcome. After aligning the outcomes, the palindromic SNPs were removed15.To eliminate weak IVs, we utilize the F-statistic to verify that the genetic instruments for exposure possess adequate strength. Biases caused by weak IVs in a two-sample model can be effectively avoided only if the F-statistic exceeds 1016.
To assess the causal effects of the gut microbiome and inflammatory proteins on arthritis, we conducted separate two-sample MR analyses (as illustrated in Phases 1 A and 2 A of Fig. 1). The inverse variance-weighted (IVW) method served as the primary analytical approach, and the Wald ratio test was applied to traits with only one IV17. MR results were presented as odds ratios with corresponding 95% confidence intervals. The results were considered statistically significant when the P-value of the IVW method was less than 0.05 and the directions of the IVW and MR-Egger were consistent. The IVW method is a standard tool in MR analysis, widely applied in multi-instrument variable settings due to its ability to maximize statistical efficiency. By weighting each SNP’s effect estimate inversely to its variance, IVW integrates information from multiple SNPs, ensuring a more precise estimation18. Compared to the simple mode method, IVW not only provides higher statistical power but also minimizes the influence of weak instrumental variables by properly weighting instruments based on their effect sizes, resulting in more robust and reliable findings.
Based on the two-sample MR analysis phases outlined in Fig. 1 (Phases 1 A and 2 A), the gut microbiota and inflammatory proteins that demonstrated significant causal effects on arthritis were included in the mediation analysis. We investigated whether the gut microbiota exerts a causal influence on inflammatory proteins (Fig. 1; Phase 3, Path a). After confirming their causal effects, we explored the mediating role of inflammatory proteins in the pathway from gut microbiota to arthritis through multiple rounds of MR analysis. The mediation effect calculation primarily used the coefficient method, and the delta method was employed to estimate the specific proportion of the mediation effect19.
To explore the bidirectional causal relationships among the gut microbiome, inflammatory proteins, and arthritis, we treated arthritis as the “exposure” and the associated gut microbiota or inflammatory proteins as the “outcomes” (as indicated in Phases 1B and 2B in Fig. 1). We selected SNPs that were significantly associated with arthritis (P < 5 × 10–8) as the IVs.
To evaluate the heterogeneity of each SNP, we employed Cochran’s Q test and visually represented the MR analysis results as scatter plots depicting the SNP-exposure and SNP-outcome relationships20. We conducted a leave-one-out analysis to explore the influence of each SNP on the study outcomes. This was performed through sequentially excluding each SNP and applying the IVW method to assess the impact of the remaining SNPs on the specific variant estimates. Additionally, we used MR-PRESSO and MR-Egger regression to detect potential issues associated with horizontal pleiotropy. MR-PRESSO addressed horizontal pleiotropy by identifying and excluding outliers21.
To investigate the role of the gut microbiota in the development and mechanisms of arthritis, we selected genes near key SNPs that were identified in the MR analysis from Phases 1 A for GO and KEGG pathway enrichment analyses (Phases 4). Using R statistical software, these key SNPs were subjected to GO analyses of their cellular composition, molecular functions, and biological processes, along with KEGG pathway enrichment analysis. We considered the results to be statistically significant when the P-value was less than 0.05.
All analyses were conducted using R statistical software (version 4.2.1, R Foundation for Statistical Computing, Vienna, Austria). The “TwoSampleMR” package, which is based on R, was used for conducting the MR analyses. The “MR_PRESSO” package was used for multiplicity testing.
Results
Genetic instrument selection
First, we identified 1496, 434, 280, 224, and 125 SNPs associated with 195 gut microbiota at the genus, family, order, class, and phylum levels, respectively, at a significance level of P < 1 × 10−5 (16 gut microbiota were excluded because they did not meet the screening criteria). These 2559 SNPs were selected as IVs for the 195 gut microbiota taxa (Supplementary File 1: Table S1). Subsequently, we identified 1820 SNPs associated with 91 inflammatory proteins at a significance level of P < 5 × 10−6 (Supplementary File 1: Table S2).
Causal influence of gut microbiota and inflammatory proteins on various types of arthritis
Osteoarthritis
As shown in Fig. 2, the MR analysis indicated that genetic predictions for the two types of gut microbiota, family Actinomycetaceae (OR = 1.086, 95% CI = 1.001–1.179, P = 0.048) and order Actinomycetales (OR = 1.093, 95% CI = 1.010–1.184, P = 0.028), were associated with an increased risk of OA (Supplementary File 2: Table S1).
The two genera of gut microbiota, Desulfovibrio (OR = 0.934, 95% CI = 0.873–0.999, P = 0.048) and Parasutterella (OR = 0.924, 95% CI = 0.858–0.995, P = 0.037) were predicted to be associated with a reduced risk of OA (Supplementary File 2: Table S1).
As shown in Fig. 3, fractalkine (OR = 1.070, 95% CI = 1.016–1.126, P = 0.01) and STAM-binding protein (OR = 1.093, 95% CI = 1.010–1.184, P = 0.028) measurements significantly increased the risk of OA. Meanwhile, the level of neurturin in blood plasma (OR = 0.932, 95% CI = 0.878–0.988, P = 0.019) measurements decreased the risk of OA onset (Supplementary File 3: Table S1).

Mendelian randomization results of the causal effects between the gut microbiota and four types of arthritis.
Rheumatoid arthritis
As shown in Fig. 2, the MR analysis demonstrated that genetic predictions of the three types of gut microbiota, genus Butyricimonas (OR = 1.133, 95% CI = 1.009–1.272, P = 0.034), genus Ruminococcaceae UCG002 (OR = 1.163, 95% CI = 1.039–1.303, P = 0.009), and order Mollicutes RF9 (OR = 1.185, 95% CI = 1.067–1.315, P = 0.002), were associated with an increased risk of RA (Supplementary File 2: Table S2).
Moreover, genetic predictions of the five types of gut microbiota, family Acidaminococcaceae (OR = 0.841, 95% CI = 0.732–0.966, P = 0.014), genus Prevotella 7 (OR = 0.892, 95% CI = 0.831–0.956, P = 0.001), genus Prevotella 9 (OR = 0.884, 95% CI = 0.800–0.976, P = 0.015), genus Sellimonas (OR = 0.919, 95% CI = 0.858–0.983, P = 0.015), and order Lactobacillales (OR = 0.849, 95% CI = 0.742–0.971, P = 0.017), were associated with a decreased risk of RA (Supplementary File 2: Table S2).
As shown in Fig. 3, several biomarker measurements were associated with changes in the risk of RA. Measurements of CD40 (OR = 1.141, 95% CI = 1.082–1.204, P < 0.001), IL-10 receptor subunit beta (OR = 1.057, 95% CI = 1.005–1.112, P = 0.032), and IL-7 (OR = 1.246, 95% CI = 1.097–1.416, P < 0.001) showed that these were associated with an increased risk of RA. In contrast, C − C motif chemokine 23 (OR = 0.920, 95% CI = 0.869–0.974, P = 0.004), fibroblast growth factor 19 (OR = 0.905, 95% CI = 0.829–0.987, P = 0.024), and IL-6 (OR = 0.874, 95% CI = 0.771–0.991, P = 0.035) were associated with a decreased risk of developing RA (Supplementary File 3: Table S2).

Mendelian randomization results of the causal effects between inflammatory proteins and four types of arthritis.
Psoriatic arthritis
As illustrated in Fig. 2, MR analysis suggested that genetic predictions of the three genera of gut microbiota, Howardella (OR = 1.151, 95% CI = 1.011–1.310, P = 0.017), Methanobrevibacter (OR = 1.260, 95% CI = 1.043–1.523, P = 0.032), and Sutterella (OR = 1.268, 95% CI = 1.002–1.603, P = 0.048), were associated with an increased risk of PsA (Supplementary File 2: Table S3).
Conversely, genetic predictions of the four types of gut microbiota, class Bacteroidia (OR = 0.719, 95% CI = 0.519–0.994, P = 0.046), family Rikenellaceae (OR = 0.715, 95% CI = 0.555–0.921, P = 0.009), Eubacterium brachy group (OR = 0.858, 95% CI = 0.741–0.993, P = 0.040), and order Bacteroidales (OR = 0.719, 95% CI = 0.519–0.994, P = 0.046), were associated with a decreased risk of PsA (Supplementary File 2: Table 3 S).
As shown in Fig. 3, certain biomarker measurements were associated with changes in the risk of PsA. Measurements of interferon-gamma (OR = 1.261, 95% CI = 1.002–1.585, P = 0.048), IL-10 receptor subunit beta (OR = 1.127, 95% CI = 1.008–1.260, P = 0.035), and TNF-related apoptosis-inducing ligand (OR = 1.123, 95% CI = 1.012–1.245, P = 0.028) indicated that these increased the risk of PsA. In contrast, IL-6 (OR = 0.772, 95% CI = 0.623–0.956, P = 0.018), and monocyte chemotactic protein 3 (OR = 0.807, 95% CI = 0.688–0.948, P = 0.009) decreased the risk of developing PsA (Supplementary File 3: Table S3).
Ankylosing spondylitis
As illustrated in Fig. 2, MR analysis indicated that genetic predictions for the three types of gut microbiota, class Actinobacteria (OR = 1.254, 95% CI = 1.004–1.566, P = 0.046), order Bacillales (OR = 1.199, 95% CI = 1.030–1.394, P = 0.019), and phylum Verrucomicrobia (OR = 1.288, 95% CI = 1.025–1.619, P = 0.030), were associated with an increased risk of AS (Supplementary File 2: Table S4).
Furthermore, genetic predictions for the three other types of gut microbiota, family Bacteroidaceae (OR = 0.660, 95% CI = 0.466–0.933, P = 0.019), genus Bacteroides (OR = 0.660, 95% CI = 0.466–0.933, P = 0.019), and genus Oscillospira (OR = 0.735, 95% CI = 0.564–0.957, P = 0.022), were associated with a reduced risk of AS (Supplementary File 2: Table S4).
As shown in Fig. 3, measurements of IL-7 (OR = 1.322, 95% CI = 1.004–1.741, P = 0.046) indicated that these significantly increased the risk of AS. Conversely, measurements of C − C motif chemokine 28 (OR = 0.796, 95% CI = 0.650–0.975, P = 0.027) and IL-6 (OR = 0.710, 95% CI = 0.515–0.977, P = 0.036) revealed that these reduced the risk of AS (Supplementary File 3: Table S4).
As shown in Fig. 4, the heatmap illustrates the causal relationships between the gut microbiota and four types of arthritis.

Heatmap depicting the causal relationships between the gut microbiota and four types of arthritis.
Sensitivity analyses
Analysis of the MR-Egger regression intercept confirmed that pleiotropy did not skew the study results. Further, the MR-PRESSO analysis results showed no significant horizontal pleiotropy in the MR studies (P > 0.05; Supplementary File 4). Cochran’s Q test did not detect any significant heterogeneity (P > 0.05; Supplementary File 4). The robustness of the MR analysis was further supported by a leave-one-out analysis in which the confidence intervals for all SNPs did not include zero (Supplementary File 5). Scatter plots were used to illustrate the comprehensive impact of the gut microbiota and inflammatory proteins on arthritis (Supplementary File 5), and forest plots were used to demonstrate their causal relationships with arthritis (Supplementary File 5).
Impact of arthritis on the gut microbiome and inflammatory proteins in reverse causality
As shown in Supplementary Files 6, no reverse interactions were observed between the gut microbiome and inflammatory proteins related to different types of arthritis, including OA, RA, and AS. The gut microbiota showed no reverse causal effect on PsA. However, there was bidirectional causal relationships between PsA and levels of the inflammatory proteins IL-6. Specifically, PsA was associated with an increased genetic risk for IL-6 (OR = 1.022, 95% CI = 1.001–1.043, P = 0.042).
Mediation analysis
This study established that both the gut microbiome and inflammatory proteins causally influence arthritis. A prerequisite for mediation is a notable correlation between the gut microbiome and inflammatory proteins. However, our findings revealed no causal links between the gut microbiome and inflammatory proteins linked to OA, RA, and PsA (Phases 3a in Fig. 1; Supplementary File 7). This suggests that inflammatory proteins do not serve as mediators in the pathway from the gut microbiome to OA, RA, and PsA. In contrast, we found a causal relationship between the order Bacillales in the gut microbiome and IL-7 in inflammatory proteins related to AS. Thus, the mediation effect of IL-7 in the pathway from the gut microbiota to AS, as calculated using the product of coefficients method, was β = 0.025 (95% CI = 0.001–0.064), accounting for 13.8% of the observed effect (Table 1).
Gene ontology and KEGG pathway enrichment analyses
As shown in Fig. 5, during GO term enrichment analysis, we identified nine biological processes related to OA. These processes primarily involved the metabolism of various nucleotides and their derivatives, including the metabolic pathways of common nucleotides, nucleotide phosphates, organic phosphate compounds, and carbohydrate derivatives. For RA, we identified 10 associated GO biological processes, including the differentiation of cardiomyocytes and striated muscle cells, development of cardiac and muscle tissues, and organization of synapses in the nervous system. In the PsA analysis, we determined 10 relevant GO biological processes involving various intracellular activities, such as phosphatidylinositol kinase and glycosyltransferase activities, activity of transcription factors and nuclear receptors, and protein palmitoylation. For AS, the analysis revealed 10 related GO biological processes that mainly focused on regulation of the clearance of low-density lipoprotein (LDL) particles, cholesterol balance, and processes related to chromosomal structure.

Gene ontology enrichment analysis scatter plots of the gut microbiota on four types of arthritis.
As shown in Fig. 6, in the KEGG pathway enrichment analysis for OA, we identified 10 related pathways, including the citric acid cycle; biosynthesis of pantothenate and its coenzyme A; β-alanine, propionate, and pyrimidine metabolism; and the biosynthesis of nucleotide sugars. However, our analysis did not reveal any significant KEGG pathway enrichment pathways associated with the gut microbiome-enriched genes in RA, PsA, and AS.

KEGG pathway enrichment analysis scatter plots of the gut microbiota on osteoarthritis.
Discussion
The concept of the gut-joint axis reveals complex interactions between the gut microbiota and joint health. This axis suggests that changes in the gut microbiota not only affect the health of the gut but may also have long-distance effects on joints22. The composition and function of both gut and oral microbiota significantly impact bone health and diseases23. Particularly in patients with RA and OA, significant changes in the gut and oral microbiota have been observed, similar to those identified in systemic inflammatory conditions, such as inflammatory bowel disease, spinal arthritis, and psoriasis, including reduced microbial diversity and disrupted immunoregulatory properties24. Age-related changes in the gut microbiota, such as reduced microbial diversity, a decrease in dominant species, and an increase in subdominant species, may play key roles in the development and progression of arthritis25.
The gut microbiota is closely associated with the host’s immune system. Gut microbes can influence the production of inflammatory proteins through various mechanisms. For example, certain gut microbes can promote the production of anti-inflammatory cytokines, such as IL-10 and TGF-β, which help to maintain immune balance and prevent excessive inflammation26. Conversely, an imbalance in the gut microbiota (dysbiosis) can lead to an increase in pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, thereby triggering or exacerbating the inflammation process. Inflammatory proteins, including both pro- and anti-inflammatory cytokines, directly participate in the pathogenesis of arthritis27. In OA and RA, the upregulation of pro-inflammatory cytokines can promote inflammatory responses within joints, leading to damage in cartilage and other joint tissues28,29.
The association between the gut microbiota and various human diseases is increasingly acknowledged. However, it remains largely unresolved whether these microbiota changes are the causes or results of diseases, or merely bystanders coincidentally affected by a third factor involved in these diseases. Walter emphasized the importance of adopting more rigorous and critical methods in the experimental design and statistical analyses used to deduce causality30. This is crucial for preventing misconceptions and unrealistic expectations that could compromise the credibility of microbiome science and delay its practical application. Distinguishing whether alterations in gut microbiota directly cause disease states or are simply associated with them is particularly important. Therefore, research designs that go beyond mere correlations are required to establish causality. In this context, methods such as MR have been used to analyze the associations between genetic data and gut microbiome composition, eliminating confounding factors to explore potential causal relationships between the gut microbiota and arthritis7. In the present study, we examined the relationship between the abundance of 211 common gut microbial populations and four types of arthritis (OA, RA, PsA, and AS). The results indicated that certain gut microbial populations are risk factors for each subtype of arthritis, whereas others may serve as protective factors.
A high abundance of the family Actinomycetaceae and order Actinomycetales was associated with an increased risk of OA. Some members of these gut microbiota were associated with bone mineral density, which is closely related to bone health31. The genera, Desulfovibrio and Parasutterella, reduced the risk of OA. Bacteria of the genus Desulfovibrio produce hydrogen sulfide (H2S) by reducing sulfates, and H2S can reduce inflammatory responses by inhibiting the production of specific inflammatory mediators, which may be a potential pathway for reducing the risk of OA32,33. Parasutterella can exert anti-inflammatory effects by producing short-chain fatty acids (SCFAs), and previous studies have found that an increase in Parasutterellaabundance is directly associated with reduced LDL levels, providing a hypothesis for its impact on OA through lipid metabolism34,35.
Genetic predictions related to the genus Butyricimonas, genus Ruminococcaceae UCG002, and order Mollicutes RF9 were associated with an increased risk of RA. Previous studies have found that autoantigens in patients with RA share sequence homology with proteins from the commensal bacterium, Butyricimonas, suggesting that the gut microbiota may trigger autoimmune responses in RA36. ELISpot assays revealed that patients with RA exhibit T-cell reactivity to these autoantigens and the corresponding microbial peptides, emphasizing the role of the gut microbiota in the pathogenesis of RA36,37. Mollicutes RF9, a class of bacteria lacking cell walls, has been less studied, and thus its pathogenic role in RA merits further investigation38. Genetic predictions related to the family Acidaminococcaceae, genera Prevotella 7, Prevotella 9, and Sellimonas, and order Lactobacillales were associated with a reduced risk of RA. In individuals with restored gut homeostasis, an increase in Sellimonas intestinalisabundance highlights its potential positive role in maintaining gut health, including its involvement in crucial metabolic processes. Additionally, the pathogenic factors and antibiotic resistance genes possessed by this strain may help protect gut microbiome balance under antibiotic interference, potentially reducing the risk of RA by maintaining gut health39. Limosilactobacillus reuteri, a member of the order Lactobacillales, exhibits antibacterial and anti-inflammatory effects by regulating the gut microbiota, host epithelial cells, immune cells, etc., offering potential therapeutic effects for various immune-related diseases, such as atopic dermatitis and RA40. The order Lactobacillales, especially lactobacilli, are a class of probiotics that play a supportive role in maintaining human intestinal health, immune function, and metabolic regulation and have great potential in the treatment of RA41.
High abundance of the genera, Howardella, Methanobrevibacter, and Sutterella, could increase the risk of PsA. Studies have shown that methane-producing archaea, such as Methanobrevibacter smithii, may affect immune responses by activating human dendritic cells and initiating innate immune responses, such as cytokine production42. Owing to limited research, the mechanisms by which they increase the risk of PsA require further investigation. Genetic predictions related to the class Bacteroidia, family Rikenellaceae, Eubacterium brachy group, and order Bacteroidales were associated with a reduced risk of PsA. Bacteroidia play a key role in maintaining intestinal and overall immune health, including the activation of T-cell responses and the regulation of pathogens in the gut. Specifically, Bacteroides thetaiotaomicronpromotes the production of the immunoregulatory cytokine, IL-10, through its outer membrane vesicles, displaying complex mutualistic interactions with the host, providing a potential route for reducing PsA43,44. In a study by Hidalgo-Cantabrana et al., individuals with psoriasis had a lower relative abundance of the family Rikenellaceae than did healthy individuals45. Rikenellaceae, by producing SCFAs and other metabolic products, can promote immune tolerance and anti-inflammatory responses, helping prevent inflammatory bowel and other immune-mediated diseases, which could be employed as a potential means of reducing the risk of PsA46.
Genetic predictions related to the class Actinobacteria, order Bacillales, and phylum Verrucomicrobia were associated with an increased risk of AS. Studies have shown that compared to that of healthy controls, the gut microbial community structure of patients with AS shows significant differences, with an observed increase in the abundance of Actinobacteria at both the genetic and species levels, especially for bacteria belonging to the genus Bifidobacterium. These bacteria are commonly used as probiotics; however, studies have suggested that some Bifidobacteriumspecies may play a role in the pathogenesis of autoimmune diseases by inducing Th2-driven immune responses47,48. Another MR study showed a positive correlation between Bacillales and the risk of AS, which is consistent with our results49. Compared to that in healthy controls, Verrucomicrobia also showed an increased relative abundance in patients with AS and correlated positively with Erythrocyte Sedimentation Rate [ESR] and C-Reactive Protein [CRP] levels50,51. Genetic predictions related to the family Bacteroidaceae, genus Bacteroides, and genus Oscillospira, however, were associated with a reduced risk of AS. Previous studies have found that the number of Bacteroidesspecies in the gut microbiome of patients with AS is significantly reduced, consistent with the reduction observed in inflammatory bowel disease5,43. In another study, besides a reduced relative abundance in patients with AS, Bacteroidesalso correlated positively with ESR and CRP levels50. Previous research suggests that moxibustion may positively affect patients with AS by increasing the abundance of the genus Oscillospira, thereby adjusting the gut microbiome structure, showing a negative correlation with human leukocyte antigen-B2752.
This study used the relative abundance of the gut microbiota to evaluate their potential impact on arthritis. However, the exact mechanisms by which the gut microbiome leads to arthritis remain unclear. We hypothesize that inflammatory proteins may act as mediators between the gut microbiome and arthritis. Therefore, exploring the interactions between inflammatory proteins and the gut microbiome could help improve our understanding of this complex association and provide more insight into the treatment of arthritis.
Two-step MR analyses revealed a causal relationship between the order Bacillales in the gut microbiome and IL-7 in the inflammatory proteins associated with AS. In the intestines of patients with AS, IL-7 levels are significantly increased, especially around the Paneth cells, where c-kit/IL-7R(+) cells accumulate. Moreover, mucosal-associated invariant T (MAIT) cells can be activated by IL-7, promoting the production of IL-17, which plays a crucial role in activating the Th17 axis in AS53,54. The Th17 axis plays an important role in AS pathology and is involved in promoting inflammation and bone proliferation. Therefore, we speculate that enrichment of Bacillales in the gut microbiome leads to an increase in IL-7 levels in the gut, which in turn activates MAIT cells and the production of IL-17. This could have a profound impact on inflammatory responses and bone pathology in the pathophysiology of AS through activation of the Th17 axis.
Moreover, arthritis itself may influence changes in the gut microbiome and inflammatory proteins. Therefore, we explored the reverse causal effects of the four arthritis subtypes on corresponding arthritis-related gut microbiomes and inflammatory proteins. Reverse MR results showed no reverse interactions between the gut microbiomes related to various types of arthritis (OA, RA, and AS) and inflammatory proteins. There was no reverse interaction observed between the gut microbiome and PsA, whereas a bidirectional causal relationship between the inflammatory proteins, IL-6, and PsA were observed.
Through GO analysis, we found that the gut microbiota was significantly different in biological processes related to the four types of arthritis investigated, emphasizing the specific roles that the gut microbiota may play in different arthritis pathologies. For example, the analysis of OA highlighted the importance of the metabolism of nucleotides and their derivatives, suggesting that these metabolic pathways may be key factors in the development of OA55,56. In contrast, the biological processes related to RA, PsA, and AS involved muscle tissue development, intracellular signaling mechanisms, and processes related to chromosomal structure, which may reflect the immune modulation characteristics of these diseases. KEGG pathway enrichment analysis unraveled the potential mechanisms by which the gut microbiota could influence arthritis through metabolic pathways. Particularly in OA, the identified pathways, such as the citric acid cycle and β-alanine metabolism, suggest that the gut microbiota might affect disease progression by influencing host energy metabolism and inflammatory responses57. However, our study did not identify any significant KEGG pathways related to genes enriched in the gut microbiota of RA, PsA, and AS, which may indicate that the influence of these pathways on these diseases are more complex or do not fully correspond to information in the current KEGG pathway database. The complex and differentiated roles that the gut microbiota may play in the development of arthritis should prompt future studies to comprehensively understand how the gut microbiota affects the development of various types of arthritis through specific biological processes and metabolic pathways, which may provide a basis for the development of new treatment strategies.
The main limitations encountered in this study include the following. First, due to the small sample size of the gut microbiome and inflammatory protein data, the accuracy of the analysis may have been affected. Second, the GWAS data analysis of the gut microbiome only covered taxonomic levels from phylum to genus and did not include more detailed species- or strain-level data. Third, the samples from the MiBioGen gut microbiome data consortium were mainly of European ancestry, which may limit the applicability of the study results across different races globally. Lastly, based on previous research experience, and due to the limited number of available SNPs, we adopted a higher threshold (1 × 10−5) in our MR analysis rather than the standard (5 × 10−8)58. However, to reduce the bias caused by weak IVs, we excluded SNPs with an F-statistic less than 10 to ensure the robustness of the analysis16.
In MR analysis, we assumed that the genetic variants used act as instrumental variables, which are valid only if they influence the outcome solely through the exposure of interest—gut microbiota and inflammatory proteins. One key MR assumption is the absence of horizontal pleiotropy, where genetic variants affect the outcome via pathways unrelated to the exposure. Pleiotropy can introduce bias and undermine causal inference. To address this, we used sensitivity analyses, including MR-Egger regression and weighted median estimation, to detect and mitigate potential pleiotropic effects, helping assess the robustness of our findings59. Horizontal pleiotropy tests showed no strong evidence for directional pleiotropy, suggesting our results are reliable. However, MR cannot fully eliminate residual confounding. Genetic variants may also not capture the dynamic, individualized nature of gut microbiota, influenced by factors like diet, antibiotics, and environment60,61. Future studies should integrate genetic, environmental, and lifestyle data for a more comprehensive understanding. Arthritis, particularly autoimmune forms like RA, is influenced by a complex interplay of genetic, environmental, and lifestyle factors62. Focusing solely on gut microbiota may oversimplify arthritis pathogenesis. Future research should explore interactions between microbiota and other factors, such as immune response and lifestyle, to better understand disease progression. Even if a causal link between gut microbiota and arthritis is established, translating these findings into effective therapies is challenging. Current interventions, such as probiotics, prebiotics, or fecal microbiota transplantation, yield inconsistent results across conditions63. The effectiveness of these approaches may depend on individual microbiota composition and host factors, highlighting the need for personalized interventions that consider genetic, microbiota, and environmental influences.
Despite these limitations, this study had several key strengths. This is the first large-scale MR analysis that explored the causal relationships between the gut microbiome, inflammatory proteins, and various subtypes of arthritis. With access to extensive public GWAS data, the accuracy and applicability of our results were significantly enhanced. Additionally, by employing an MR study design, we effectively addressed the selection bias and reverse causality issues commonly encountered in traditional epidemiological studies. We also conducted a mediation analysis to enhance the stability and replicability of our results, which enabled us to accurately identify the significant roles of the order Bacillales and IL-7 in relation to AS.
Conclusion
This study extensively explored the causal relationships between the gut microbiota, inflammatory proteins, and various forms of arthritis. We identified 11 positive and 14 negative causal associations between genetic liabilities of the gut microbiota and arthritis. In addition, we identified 9 positive and 8 negative causal associations between inflammatory proteins and arthritis. Furthermore, IL-7 strongly correlated with AS and likely mediates 13.8% of the effect exerted by the gut microbiota on AS, suggesting that modulating the gut microbiota to influence inflammatory proteins could be an effective therapeutic strategy for arthritis. Specifically, the mediation of IL-7 between gut microbiota changes and AS underscores a promising therapeutic pathway. Overall, this strategy supports the development of targeted therapies that alter the microbiome to modulate inflammatory responses and alleviate symptoms of arthritis.
Data availability
The GWAS summary statistics were obtained from https://mibiogen.gcc.rug.nl/, https://r10.risteys.finngen.fi/ and https://gwas.mrcieu.ac.uk/. All extracted data used for the analyses are included in this article and the Supplementary Information files.
Abbreviations
- AS:
-
ankylosing spondylitis
- CI:
-
confidence interval
- CRP:
-
C-Reactive Protein
- ESR:
-
Erythrocyte Sedimentation Rate
- GO:
-
gene ontology
- GWAS:
-
genome-wide association study
- H2S:
-
hydrogen sulfide
- IL:
-
interleukin
- IV:
-
instrumental variable
- IVW:
-
inverse variance-weighted
- LD:
-
linkage disequilibrium
- LDL:
-
low-density lipoprotein
- MAIT:
-
mucosal-associated invariant T
- MR:
-
Mendelian randomization
- OA:
-
osteoarthritis
- OR:
-
odds ratio
- PsA:
-
psoriatic arthritis
- RS:
-
rheumatoid arthritis
- SCFA:
-
short-chain fatty acid
- TNF:
-
tumor necrosis factor
References
Shi, Y., Wang, G., Lau, H. C. H. & Yu, J. Metagenomic sequencing for microbial DNA in human samples: emerging Technological advances. Int. J. Mol. Sci. 23, 2181 (2022).
Mousa, W. K., Chehadeh, F. & Husband, S. Recent advances in understanding the structure and function of the human microbiome. Front. Microbiol. 13, 825338 (2022).
Zheng, W. et al. High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome. Science. 376, eabm1483 (2022).
Simpkins, D. A. et al. Consequences of collagen induced inflammatory arthritis on circadian regulation of the gut microbiome. FASEB J. Off Publ Fed. Am. Soc. Exp. Biol. 37, e22704 (2023).
Wen, C. et al. Quantitative metagenomics reveals unique gut microbiome biomarkers in ankylosing spondylitis. Genome Biol. 18, 142 (2017).
Dong, Y. et al. Relationship between gut Microbiota and Rheumatoid Arthritis: A Bibliometric Analysis (Front Immunol [Internet], 2023).
Davey Smith, G. & Hemani, G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23, R89–98 (2014).
Lawlor, D. A., Harbord, R. M., Sterne, J. A. C., Timpson, N. & Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
Belizário, J. E. & Faintuch, J. Microbiome and Gut Dysbiosis. In: (eds Silvestre, R. & Torrado, E.) Metab Interact Infect [Internet]. Cham: Springer International Publishing; 459–476. (2018).
Role of Inflammatory Cytokines in the Pathogenesis. Rheumatoid Arthritis Novel Therapeutic Targets ;37–46. (2020).
Bowden, J. & Holmes, M. V. Meta-analysis and mendelian randomization: a review. Res. Synth. Methods. 10, 486–496 (2019).
Kurilshikov, A. et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165 (2021).
Zhao, J. H. et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat. Immunol. 24, 1540–1551 (2023).
Tachmazidou, I. et al. Identification of new therapeutic targets for osteoarthritis through genome-wide analyses of UK Biobank data. Nat. Genet. 51, 230–236 (2019).
Myers, T. A., Chanock, S. J. & Machiela, M. J. LDlinkR: an R Package for rapidly calculating linkage disequilibrium statistics in diverse populations. Front. Genet. 11, 157 (2020).
Pierce, B. L., Ahsan, H. & Vanderweele, T. J. Power and instrument strength requirements for mendelian randomization studies using multiple genetic variants. Int. J. Epidemiol. 40, 740–752 (2011).
Burgess, S., Dudbridge, F. & Thompson, S. G. Combining information on multiple instrumental variables in mendelian randomization: comparison of allele score and summarized data methods. Stat. Med. 35, 1880–1906 (2016).
Lin, Z., Deng, Y. & Pan, W. Combining the strengths of inverse-variance weighting and Egger regression in mendelian randomization using a mixture of regressions model. PLOS Genet. 17, e1009922 (2021).
Burgess, S., Daniel, R. M., Butterworth, A. S., Thompson, S. G. & the EPIC-InterAct Consortium. Network mendelian randomization: using genetic variants as instrumental variables to investigate mediation in causal pathways. Int. J. Epidemiol. 44, 484–495 (2015).
Cohen, J. F. et al. Cochran’s Q test was useful to assess heterogeneity in likelihood ratios in studies of diagnostic accuracy. J. Clin. Epidemiol. 68, 299–306 (2015).
Verbanck, M., Chen, C-Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Romero-Figueroa, M. D. S., Ramírez-Durán, N., Montiel-Jarquín, A. J. & Horta-Baas, G. Gut-joint axis: gut dysbiosis can contribute to the onset of rheumatoid arthritis via multiple pathways. Front. Cell. Infect. Microbiol. 13, 1092118 (2023).
Drago, L. et al. Oral–gut microbiota and arthritis: is there an evidence-based Axis? J. Clin. Med. 8, 1753 (2019).
Hao, X. et al. The gut microbiota in osteoarthritis: where do we stand and what can we do? Arthritis Res. Ther. 23, 42 (2021).
Golovach, I. Y. & Rekalov, D. G. Osteoarthritis and intestinal microbiota: pathogenetic significance of the joint — gut — microbiome axis. PAIN Jt. SPINE. 12, 72–80 (2022).
Halami, H. S. R. The combined effect of potential probiotic Bacillus licheniformis MCC 2514 and Bifidobacterium breve NCIM 5671 towards anti-inflammatory activity on HT-29 cell lines. Probiotics Antimicrob. Proteins. 15, 351–362 (2023).
Chow, Y. Y. & Chin, K-Y. The role of inflammation in the pathogenesis of Osteoarthritis. Mediators Inflamm. 2020, e8293921 (2020).
Jrad, A. I. S., Trad, M., Bzeih, W., El Hasbani, G. & Uthman, I. Role of pro-inflammatory interleukins in osteoarthritis: a narrative review. Connect. Tissue Res. 64, 238–247 (2023).
Alunno, A., Carubbi, F., Giacomelli, R. & Gerli, R. Cytokines in the pathogenesis of rheumatoid arthritis: new players and therapeutic targets. BMC Rheumatol. 1, 3 (2017).
Walter, J., Armet, A. M., Finlay, B. B. & Shanahan, F. Establishing or exaggerating causality for the gut microbiome: lessons from Human Microbiota-Associated rodents. Cell. 180, 221–232 (2020).
Chen, S., Zhou, G., Han, H., Jin, J. & Li, Z. Causal effects of specific gut microbiota on bone mineral density: a two-sample mendelian randomization study. Front. Endocrinol. 14, 1178831 (2023).
Baumgartner, L. K. et al. Sulfate reducing bacteria in microbial mats: changing paradigms, new discoveries. Sediment. Geol. 185, 131–145 (2006).
Pan, L-L., Qin, M., Liu, X-H. & Zhu, Y-Z. The role of Hydrogen Sulfide on Cardiovascular Homeostasis: an overview with update on Immunomodulation. Front. Pharmacol. 8, 686 (2017).
Shin, Y. et al. Roles of short-chain fatty acids in inflammatory bowel disease. Nutrients. 15, 4466 (2023).
Henneke, L. et al. A dietary carbohydrate - gut Parasutterella - human fatty acid biosynthesis metabolic axis in obesity and type 2 diabetes. Gut Microbes. 14, 2057778 (2022).
Pianta, A. et al. Two rheumatoid arthritis–specific autoantigens correlate microbial immunity with autoimmune responses in joints. J. Clin. Invest. 127, 2946–2956 (2017).
Coradduzza, D. et al. Decoding the Microbiome’s influence on rheumatoid arthritis. Microorganisms. 11, 2170 (2023).
Chernova, O. A. et al. Antimicrobial drug resistance mechanisms among Mollicutes. Int. J. Antimicrob. Agents. 57, 106253 (2021).
Muñoz, M. et al. Comprehensive genome analyses of Sellimonas Intestinalis, a potential biomarker of homeostasis gut recovery. Microb. Genomics. 6, mgen000476 (2020).
Luo, Z. et al. Limosilactobacillus reuteri in immunomodulation: molecular mechanisms and potential applications. Front. Immunol. 14, 1228754 (2023).
Rastogi, S. & Singh, A. Gut microbiome and human health: exploring how the probiotic genus Lactobacillus modulate immune responses. Front. Pharmacol. 13, 1042189 (2022).
Bang, C., Weidenbach, K., Gutsmann, T., Heine, H. & Schmitz, R. A. The intestinal Archaea Methanosphaera stadtmanae and Methanobrevibacter smithii activate human dendritic cells. PLOS ONE. 9, e99411 (2014).
Durant, L. et al. Bacteroides thetaiotaomicron-derived outer membrane vesicles promote regulatory dendritic cell responses in health but not in inflammatory bowel disease. Microbiome. 8, 88 (2020).
Parker, B. J., Wearsch, P. A., Veloo, A. C. M. & Rodriguez-Palacios, A. The Genus Alistipes: gut Bacteria with emerging implications to inflammation, Cancer, and Mental Health. Front. Immunol. 11, 906 (2020).
Hidalgo-Cantabrana, C. et al. Gut microbiota dysbiosis in a cohort of patients with psoriasis. Br. J. Dermatol. 181, 1287–1295 (2019).
Parada Venegas, D. et al. Short chain fatty acids (SCFAs)-Mediated gut epithelial and Immune Regulation and its relevance for inflammatory Bowel diseases. Front. Immunol. 10, 277 (2019).
Wang, L. et al. Gut microbiota changes in patients with spondyloarthritis: a systematic review. Semin Arthritis Rheum. 52, 151925 (2022).
Wang, Q. et al. Characteristics of the gut microbiome and its Relationship with Peripheral CD4 + T cell subpopulations and cytokines in rheumatoid arthritis. Front. Microbiol. 13, 799602 (2022).
Tang, J., Mo, S., Fan, L., Fu, S. & Liu, X. Causal association of gut microbiota on spondyloarthritis and its subtypes: a mendelian randomization analysis. Front. Immunol. 15, 1284466 (2024).
Liu, G., Hao, Y., Yang, Q. & Deng, S. The Association of Fecal Microbiota in Ankylosing Spondylitis cases with C-Reactive protein and erythrocyte sedimentation rate. Mediators Inflamm. 2020, 8884324 (2020).
Liu, G., Ma, Y., Yang, Q. & Deng, S. Modulation of inflammatory response and gut microbiota in ankylosing spondylitis mouse model by bioactive peptide IQW. J. Appl. Microbiol. 128, 1669–1677 (2020).
Sun, G., Wang, Q., Cao, S., Xu, H. & Zhao, Y. Governor Vessel Moxibustion Therapy Improves Microbiota Structure in Ankylosing Spondylitis Patients. Xu Y, editor. Dis Markers. ;2021:1–8. (2021).
Gracey, E. et al. IL-7 primes IL-17 in mucosal-associated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann. Rheum. Dis. 75, 2124–2132 (2016).
Ciccia, F. et al. Type 3 innate lymphoid cells producing IL-17 and IL-22 are expanded in the gut, in the peripheral blood, synovial fluid and bone marrow of patients with ankylosing spondylitis. Ann. Rheum. Dis. 74, 1739–1747 (2015).
Kobayashi, H. et al. The Nicotinamide Adenine Dinucleotide (NAD)-Dependent deacetylase Sirtuin-1 regulates Chondrocyte Energy Metabolism through the modulation of Adenosine Monophosphate-activated protein kinase (AMPK) in osteoarthritis(OA). (2017). J Arthritis [Internet].
Navarro, M. N., Gómez de Las Heras, M. M. & Mittelbrunn, M. Nicotinamide adenine dinucleotide metabolism in the immune response, autoimmunity and inflammageing. Br. J. Pharmacol. 179, 1839–1856 (2022).
Adams, S. B., Setton, L. A. & Nettles, D. L. The role of metabolomics in osteoarthritis research. J. Am. Acad. Orthop. Surg. 21, 63–64 (2013).
Gagnon, E. et al. Impact of the gut microbiota and associated metabolites on cardiometabolic traits, chronic diseases and human longevity: a mendelian randomization study. J. Transl Med. 21, 60 (2023).
Van Kippersluis, H. & Rietveld, C. A. Pleiotropy-robust mendelian randomization. Int. J. Epidemiol. 47, 1279–1288 (2018).
Leeming, E. R. et al. The complexities of the diet-microbiome relationship: advances and perspectives. Genome Med. 13, 10 (2021).
Schwartz, D. J., Langdon, A. E. & Dantas, G. Understanding the impact of antibiotic perturbation on the human microbiome. Genome Med. 12, 82 (2020).
Jiang, X. & Alfredsson, L. Modifiable environmental exposure and risk of rheumatoid arthritis—current evidence from genetic studies. Arthritis Res. Ther. 22, 154 (2020).
Zhu, Z., Zhang, J., Jiang, Z. & Lu, F. Treatment of chronic nonbacterial prostatitis by modifying gut microbiota: a step forward, a long way to go. J. Transl Med. 22, 1–3 (2024).
Acknowledgements
We would like to thank all participants and investigators of the included GWAS studies for their contributions to the data.
Funding
This study was supported by the Liaoning Provincial Science and Technology Plan (Grant No. 2022JH2/101500007).
Author information
Authors and Affiliations
Contributions
In this study, PBX and WY conceived and designed the study. PBX, GQH, CJN and CL analyzed the data and interpreted the results. PBX, SP, and WY wrote the manuscript and revised the manuscript. WY and SP reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pan, B., Guo, Q., Cai, J. et al. Investigating the causal impact of gut microbiota on arthritis via inflammatory proteins using mendelian randomization. Sci Rep 14, 27433 (2024). https://doi.org/10.1038/s41598-024-79336-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-79336-9
Keywords
This article is cited by
-
Investigating the causal relationship between the gut microbiome and rheumatoid arthritis: mediating effects of immune cells
Journal of Translational Medicine (2025)
