
Abstract
In this work, a novel series of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n as new potent α-glucosidase inhibitors was designed, synthesized, and evaluated. All the synthesized derivatives 10a-n were more potent than acarbose (positive control). Representatively, (E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-phenethylacetamide (10n), as the most potent entry, with IC50 = 48.4 µM was 15.5-times more potent than acarbose. According to kinetic study, compound 10n was a competitive inhibitor against α-glucosidase. This compound formed the desired interactions with important residues of the binding pocket of α-glucosidase with favorable binding energy in the molecular docking and molecular dynamics. Compounds 10n, 10e, and 10 g as the most potent compounds among the synthesized compounds were evaluated in term of pharmacokinetics and toxicity via online servers. These evaluations predicted that compounds 10n, 10e, and 10 g had good pharmacokinetic properties and toxicity profile.
Similar content being viewed by others
Introduction
Diabetes mellitus (DM) is the most common metabolic disorder in the world and the most common type of this disorder is type 2.1 DM had two main forms; type 1 that accomplished with low secretion of insulin and type 2 that accomplished with resistance of the body to the effect of insulin.2 The result of both types of DM is increasing in the level of glucose in the blood which, if not treated, leads to numerous complications such as nephropathy, retinopathy, cardiomyopathy, and neuropathy.3 One of the main ways to treat DM is decreasing postprandial hyperglycemia by oral drugs.4 An important mechanism for the latter drugs is inhibition of the decomposing enzymes of food carbohydrates into glucose such as α-glucosidase.5 α-Glucosidase is a small intestinal enzyme which hydrolyzes the glycosidic bonds of polysaccharides and disaccharides to monosaccharides such as glucose.6 Therefore, inhibition of this enzyme leads to maintenance of postprandial blood glucose level at a normal or near normal range.7 Four α-glucosidase inhibitors acarbose, miglitol, emiglitate, and voglibose are being clinically. The long-term use of these drug is associated with gastrointestinal side effects included diarrhea, abdominal discomfort, bloating pain, flatulence, and enteritis.8 Therefore, the development of potent and safe α-glucosidase inhibitors to treat DM is essential.
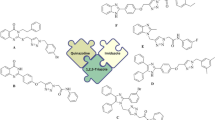
One of the ways to introduce lead drug structures in medicinal chemistry is to use molecular hybridization theory.9 Using this theory, pharmacophores are selected from biologically active compounds and connected together by common chemical reactions, to achieve more effective drugs with fewer side effects. Recently, molecular hybridization has been given much attention in the design of compounds with anti-α-glucosidase activities.10,11,12 In the present work, our research group introduced a new series of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide hybrids as the potent α-glucosidase inhibitors.
Results and discussion
Rational and design
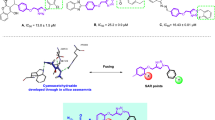
One of the popular building blocks in the design of α-glucosidase inhibitors is quinoline.13,14,15,16 As you can see in Fig. 1, even simple derivatives of quinoline such as compound A can inhibit α-glucosidase.17 On the other hand, thiosemicarbazone and 1,2,3-triazole-N-phenylacetamide pharmacophores were observed in the numerous synthetic potent α-glucosidase inhibitors such as compounds B and C.18,19 In this regards, considering the three latter pharmacophores, our research group designed the quinoline-thiosemicarbazone-1,2,3-triazole-aceamide scaffold as a new skeleton with anti-α-glucosidase property (Fig. 1). Fourteen derivatives of this scaffold were synthesized and evaluated as α-glucosidase inhibitors by in vitro and silico assessments.

Structures and inhibitory activities of the reported compounds A-C that were used in the design of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide skeleton as a new scaffold with anti-α-glucosidase property.
Chemistry
Synthesis of desired quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n was performed as outlined in Fig. 2. Required starting material, 2-(prop-2-yn-1-ylthio)quinoline-3-carbaldehyde 3 was obtained by the reaction of 2-mercaptoquinoline-3-carbaldehyde 1 and propargyl bromide 2 in the presence of K2CO3 in the acetone at room temperature. Then, reaction of compound 3 and in situ prepared azide derivatives 7a-n in the presence of catalytic amounts of CuSO4·5H2O and sodium ascorbate afforded quinoline-1,2,3-triazole-acetamide derivatives 8a-n.20 Finally, reaction of compounds 8a-n and hydrazinecarbothioamide 9 in the mixture of acetic acid and methanol provided the title compounds 10a-n.

Synthesis of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n: (a) K2CO3, Aceton, RT, 1 h; (b) DMF, RT, 1 h; (c) Et3N, H2O/t-BuOH, RT, 1 h; (d) CuSO4·5H2O, Sodium ascorbate, RT, 24–48 h; (e) Methanol, Acetic acid, Reflux, 4 h.
A proposed mechanism for the formation of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n is depicted in Fig. 3.

The proposed reaction mechanism for the formation of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n.
α-Glucosidase inhibition assay
The target compounds 10a-n were evaluated for their in vitro inhibitory activities against yeast α-glucosidase in comparison with marketed α-glucosidase inhibitor acarbose. The structures and anti-α-glucosidase activities (µM) of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n are shown in Table 1. The obtained IC50 values of test compounds 10a-n and positive control acarbose demonstrated that all new compounds were more potent than acarbose. The most active compounds were N-phenethylacetamide and N-4-ethylphenyl derivatives (compounds 10n and 10e) with IC50 values ≤ 70.3 µM (IC50 for acarbose = 750.1 µM). Furthermore, N-2,6-dimethylphenyl derivative exhibited good anti-α-glucosidase activity (IC50 value = 150.3 µM).
Structure–activity relationships of the new compounds 10a-n
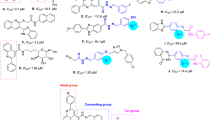
As can be seen in Fig. 2 and Table 1, fourteen derivatives 10a-n of title scaffold were synthesized. These compounds, with the exception of compound 10n, were N-phenylacetamid derivatives. Compound 10n is a N-phenethylacetamide derivative that showed highest inhibition effect among the all synthesized compounds. Interestingly, structural isomer of compound 10n, N-4-ethylphenyl derivative 10e, was the second potent compound among the synthesized compounds.
According to structures of the synthesized compounds, structure–activity relationships were evaluated on N-phenylacetamid derivatives 10a-m. As mentioned, the most potent compound among the N-phenylacetamid derivatives, was 4-ethylphenylacetamide derivative 10e. Removing of ethyl substituent and or replacement of this substituent with methyl group, as in case of compounds 10a and 10d, anti-α-glucosidase activity diminished to 5.38 and 6.97 folds, respectively. As can be seen Table 1, third potent compound was 2,6-dimethyl derivative 10g. Replacement of the second methyl substituent of 6-position to 3-position and or removing of this methyl, as in case of compounds 10f. and 10b, the inhibitory activity diminished to 1.6 and 2.3 folds, respectively. On the other hand, 3-methyl and 2,3-dimetyl derivatives 10c and 10f. showed same inhibitory activity against α-glucosidase. Introduction of a strong electron donating group such as hydroxyl group on 4-position of phenyl ring in un-substituted compound 10a slightly increased anti-α-glucosidase activity, as in case of compound 10h, while introduction of methoxy group as a moderate electron donating group in the latter position dramatically increased inhibitory activity against α-glucosidase as observed in compound 10i. On the other hand, introduction of electron withdrawing group 4-bromo or 4-nitro on phenyl ring of un-substituted compound 10a, as in case of compounds 10j and 10m, dramatically reduced anti-α-glucosidase activity. Movement of nitro group of 4-position to 3-position and or introduction of 2-methyl group in compound 10m, as in case of compounds 10k and 10l, the inhibitory activity increased to 2.58 and 1.82 folds, respectively. In general, SAR survey on anti-α-glucosidase activity of N-phenylacetamide derivatives 10a-m was schematically showed in Fig. 4.

Diagram of SAR of compounds 10a-m against α-glucosidase.
Comparison of new compounds with the used templates
The comparison of IC50 values of template compound A and compounds B1 and C1 as the most potent compounds among the template compounds B-C with the most potent compound among the new compounds 10a-n, compound 10n, revealed that our new potent compound was more potent than compound A and compound B1 but compound 10n was significantly weaker than the compound C1 (Fig. 5).17,18,19

Comparison of α-glucosidase inhibitory activity between template compounds A, B1 and C1 with the most potent new compound 10n.
Kinetic study
To further understand the α-glucosidase inhibition mechanism, kinetic study of the most potent compound 10n was performed. As can be seen in Fig. 6a, the values of Km increased with increasing concentration of the inhibitor and the values of Vmax remained the same. This finding suggested that compound 10n was a competitive inhibitor against α-glucosidase. Besides, the Ki value was determined as 48.1 µM by using Dixon plots analysis of enzymatic reactions (Fig. 6b).

(a) Lineweaver–Burk plot for compound 10n (the concentrations of compound 10n were 0, 12.1, 24.2, and 48.4 µM. (b) Secondary re-plot of slopes of Lineweaver–Burk plot vs various concentrations of the compound 10n.
Molecular modeling study on the modeled yeast α-glucosidase
To clarify the interaction modes and binding energies of the title compounds in the active site of α-glucosidase, molecular modeling study was conducted using Auto Dock Tools (version1.5.6) on the modeled yeast α-glucosidase because we used of the yeast form of this enzyme in the in vitro evaluations. Acarbose as positive control and compounds 10n, 10e, and 10 g as most potent new compounds were docked in the active site of modeled α-glucosidase.21 The superposed structure of acarbose and compound 10n (the most potent compound) is shown in Fig. 7.

Acarbose (cyan) and the most potent compound 10n (pink) superimposed in the α-glucosidase active site.
Interaction of acarbose is shown in Fig. 8. Important amino acids in active site of this enzyme are included Glu304, Thr307, Gln322, Asn241, His279, Pro309, Arg312, Thr301, and Ser308. As can be seen in Fig. 8, the thiosemicarbazone moiety of the most active compound 10n formed two hydrogen bonds with residues Pro309 and Asn241. Quinoline ring of this compound established two π-anion interactions with Glu304, a π-π interaction with His279, and a hydrophobic interaction with Pro309. Sulfur attached to quinoline ring and 1,2,3-triazol ring formed two hydrogen bonds with Arg312. The latter amino acid also established a hydrophobic interaction with 1,2,3-triazol ring. CH2 unit attached to 1,2,3-triazol ring formed a non-classical hydrogen bond with Asp408. Furthermore, N-phenethylacetamide moiety of compound 10n interacted with Phe300 via a π-π interaction.

Interaction modes of acarbose and the most potent compounds 10n, 10e, and 10 g in the active site of α-glucosidase.
The second potent compound 10e established five hydrogen bonds with residues Gln350, Glu304 (two interactions), Asp349, and Asn241 via thiosemicarbazone moiety (four interactions) and carbonyl unit of N-4-ethylphenyl acetamide moiety (one interactions). Phenyl ring of the latter moiety formed a hydrophobic interaction with Pro309. Sulfur attached to quinoline ring and 1,2,3-triazol ring of compound 10e created π-sulfur and π-π interactions with His279 and His239, respectively. Moreover, quinoline ring established a π-π interaction with Tyr313 and two hydrophobic interactions with Arg312.
The third potent compound 10g formed two hydrogen bond with active site residues Pro309 and Thr307 via thiosemicarbazone moiety and sulfur attached to quinoline ring, respectively. The latter ring established two hydrophobic interactions with Pro309. Thiosemicarbazone moiety also formed a π-sulfur interaction with His239. In addition to the mentioned interactions, 1,2,3-triazol ring of compound 10g formed a hydrophobic interaction with Arg312 and a non-classical hydrogen bond with His239. Moreover, N-2,6-dimethylphenylacetamid moiety of this compound established two hydrophobic interactions with Phe157 and His239.
The values for the binding energies (BEs) of acarbose and the compounds 10n, 10e, and 10g were − 4.04, − 8.05, − 7.91, and − 7.89 kcal/mol, respectively. Therefore, our new compounds with lower Bes in comparison to acarbose attached to the α-glucosidase’s active site easier than the latter drug. This in silico finding is in agreement with in vitro observations.
Molecular modeling study on the human α-glucosidase
As a complementary study, the most potent compound 10n was placed in the active site of a human α-glucosidase with PDB ID of 2QMJ in the protein data bank.22 In the Fig. 9, the superimpose structure of our new potent compound 10n and the positive control acarbose was shown.

Superimposed structure of acarbose (cyan) and the most potent compound 10n (pink) in the human α-glucosidase active site.
As can be seen in Fig. 10, the interaction mode of acarbose demonstrated this inhibitor established eleven hydrogen bonds with residues Asp203 (two interactions), Arg526 (two interactions), Asp54 (two interactions), Asp443 (two interactions), Gln603 (two interactions), and Tyr605 (one interaction). This drug also formed the following interactions: a non-classical hydrogen bond with Asp327, a sulfur-x interaction with Met444, a π-lone pair interaction with Tyr299, and a hydrophobic interaction with Phe575 (Fig. 10).

Interaction modes of acarbose and the most potent compound 10n in the active site of human α-glucosidase.
New potent compound 10n established the following interactions with active site of the human α-glucosidase: four classical hydrogen bonds with Gln603 (two interactions), Asp203 (one interaction), and Asp542 (one interaction), one non-classical hydrogen bond with Glu603, three π-π interactions with Tyr299, a π-sulfur interaction with Phe575, two π-anion interaction with Asp542, and an unfavorable donor-donor interaction with Arg526.
The BE values of acarbose and compound 10n in the active site of human α-glucosidase were − 5.99 and − 7.55 kcal/mol, respectively. Therefore, according to this study, it is predicted that the new compound 10n can inhibit the human α-glucosidase better than acarbose.
Molecular dynamics
The process of a substrate binding to the active site of an enzyme, like other molecular events, is a dynamic phenomenon. Therefore, simulating and analyzing enzyme–substrate complex behavior in a natural-like environment, including water and ions, can offer insights into enzyme–substrate complex stability and flexibility. In this study, the docking files of acarbose, utilized as the standard inhibitor, and compound 10n, identified as the most potent inhibitor against α-glucosidase based on in vitro studies, were subjected to molecular dynamics (MD) simulation in an explicit hydration environment. The aim was to assess the stability and flexibility of the enzyme- ligand complex over the simulation time. Two types of simulations were conducted. Initially, a 10 ns simulation was run for all complexes. It was observed that both acarbose and 10n remained stable at the α-glucosidase active site during this step. Consequently, the simulation time was extended by another 10 ns to gain deeper insights into the behavior of these compounds within the active site. The complexes were stabile throughout the extended simulation time too. Subsequently, the simulation trajectories of these compounds were subjected to further analysis using various tools. To assess the stability of the complexes, root-mean-square deviation (RMSD) and radius of gyration (Rg) calculations were conducted for all structures in the trajectory. Graphs depicting changes over time were generated using these calculations. Furthermore, the root mean square fluctuation (RMSF) of the backbone atoms in α-glucosidase and the heavy atoms in the ligands were computed to evaluate the residual flexibility of the enzyme and the flexibility of the ligand atoms, respectively.
Based on Fig. 11, the RMSD calculations indicate that α-glucosidase maintains a consistently low RMSD value, remaining below 2.5 Å throughout the entire simulation period. This trend suggests a stable protein structure. Specifically, the average RMSD values for α-glucosidase in complex with acarbose and/or 10n were 1.88 Å and 1.94 Å, respectively. Furthermore, the RMSD values of acarbose and/or 10n in complex with α-glucosidase consistently remained below 2.5 Å, with average RMSD values of 1.20 Å for acarbose and 1.47 Å for 10n. These findings indicate the presence of stable structures for both α-glucosidase and the ligands. To assess the protein’s compactness throughout the simulation, the radius of gyration (Rg) of α-glucosidase was computed (Fig. 11). The Rg values for α-glucosidase while bound to acarbose and/or 10n consistently fell within a narrow range of 23.8 to 25.4 Å, showing no significant upward or downward trends during the simulation period. The average Rg for α-glucosidase was 25.0 Å and 24.6 Å when complexed with acarbose and 10n, respectively.

Superimposed RMSD of Cα atoms of α-glucosidase in complex with 10n (blue) and acarbose (red) (A). Superimposed RMSD of 10n (blue) and acarbose (red) in complex with α-glucosidase (B). Time dependence of the radius of gyration (Rg) graph of α-glucosidase in complex with 10n (blue) and acarbose (red) (C).
In a protein, every atom and residue experiences a certain degree of fluctuation. Figure 12 illustrates the fluctuation patterns of α-glucosidase residues when bound with acarbose and 10n. According to the figure, the RMSF of α-glucosidase residues in complex with acarbose and 10n show remarkable similarity, almost identical. α-Glucosidase, a large protein consisting of 579 residues and multiple domains with unique structures and functions, displays diverse fluctuation levels across its various segments, as depicted in Fig. 12. Positioned in a cleft between the A ___domain and B ___domain, the enzyme’s active site experiences minimal fluctuation in residues engaged in non-bonded interactions with ligands. Conversely, residues located in loop regions, like the B ___domain loop and the active site lid, exhibit expectedly higher levels of fluctuation. Figure 12 depicts the fluctuation of heavy atoms in acarbose and 10n too. It’s noticeable that the RMSF of all heavy atoms in these ligands stays below 1.5 Å. This minimal fluctuation suggests a stable complex with α-Glucosidase, indicating effective constraint of their movement by intermolecular interactions.

(A) RMSF graph of the Cα atoms of α-glucosidase in complex with 10n (blue) and acarbose (red). Close-up representation of α-glucosidase active site (B). RMSF graph of the heavy atoms of 10n (C) and acarbose (D) in complex with α-glucosidase. Structure of these compounds are illustrated.
Druglikeness evaluation and ADMET prediction
Druglikeness evaluation and ADMET prediction of the positive control acarbose and the most potent new α-glucosidase inhibitors 10n, 10e, and 10 g were performed using by an online software (PreADMET) and the obtained data were listed in Table 2.23 A general method for evaluation of druglikeness is Lipinski 'Rule of five’. The obtained results of online software demonstrated that compounds 10n, 10e, and 10 g followed of this rule while acarbose violated.
ADMET represented important pharmacokinetic factors included absorption, distribution, metabolism, excretion, and toxicity. In silico prediction of these factors demonstrated that all studied compounds (acarbose, 10n, 10e, and 10 g) have poor permeability to Caco2 cell. Acarbose has not human intestinal absorption (HIA) while compounds 10n, 10e, and 10 g have high HIA. Permeability of acarbose, 10n, 10e, and 10 g to blood brain barrier (BBB) and skin is in the acceptable. In term of mutagenicity, all the latter compounds are mutagen. In term of carcinogenicity, new compounds 10n, 10e, and 10 g have not carcinogenic effect on mouse and rat while acarbose has carcinogenic effect on mouse. Finally, in term of cardiotoxicity (hERG inhibition), acarbose and compounds 10n and 10e are ambiguous and compound 10 g is high risk.
The druglikness, ADME, and medicinal chemistry criteria of the most potent compounds 10n, 10e, and 10 g were also evaluated by SwissADME online software.24 The obtained results were listed in Table 3. As can be seen in Table 3, new compounds 10n, 10e, and 10 g fallowed of Lipinski rule of five but did not fallow of Veber rule while acarbose did not fallow of these two rules. Gastrointestinal (GI) absorption of studied new compounds and acarbose was low and these compounds had not permeably to BBB. Compound 10n and acarbose were substrate for P-glycoprotein (P-gp) that is a drug efflux pump but compounds 10e and 10 g were not substrate for this pump. Cytochrome P450 (CYP) enzyme play a pivotal role in metabolism of drugs. As can be seen Table 3, compound 10n, 10e, and 10 g inhibited isoforms 2C19, 2C9, and 3A4, but they had no inhibitory effect on the 1A2 and 2D6. Acarbose did not inhibit any of the P450 isoforms. Medicinal chemistry criteria of compounds 10n, 10e, and 10 g was evaluated and obtained results was compared to acarbose (Table 3). All of the studied compounds no alerts in their pan-assay interference compounds (PAINS) descriptions, indicating that they can be suitable as drug candidates. The synthetic accessibility scores of the new compounds 10n, 10e, and 10 g were better than acarbose. This score is a metric that predicts the complexity of synthesizing drug-like molecules.
Toxicity of new compounds was also evaluated by pkCSM online software.25 AMES toxicity test of acarbose and compounds 10n, 10e, and 10 g showed that all these compounds had not carcinogen effect. The maximal tolerated dose (MTD) is important in assessing the toxicity of the new chemical compounds. MTD higher values are more desirable. MTD values for our new compounds were more than acarbose and therefore, acarbose is considered as a safer drug when compared with our new compounds in this type of toxicity. Predictions of the inhibitory effect on hERG 1 and hERG 2 indicated that our new compounds and acarbose only inhibited hERG2. Oral rat acute toxicity expressed with LD50 values and the predicted of LD50 for the selected compounds 10n, 10e, and 10 g and acarbose showed that our new compounds with more dose causes the death of 50% of test animals in comparison to acarbose. In contrast, prediction of lowest observed adverse effect level (LOAEL) that indicated oral rat chronic toxicity, demonstrated that new compounds 10n, 10e, and 10 g in lower dose created chronic toxicity in comparison to acarbose. Finally, it is predicted that new compounds 10n, 10e, and 10 g had hepatotoxicity and did not show skin sensitivity.
In vitro anti-α-amylase assay
The most potent compounds 10n, 10e, and 10 g was evaluated against pancreatic α-amylase as another important enzyme in the degradation of carbohydrates.26 As can be seen Table 3, anti-α-amylase inhibition assay of compounds 10n, 10e, and 10 g showed that these compounds with IC50 values > 200 were considered as inactive compounds against α-amylase when compared with positive control acarbose (IC50 = 108 ± 0.71 µM).
Experimental
Synthesis of 2-(prop-2-yn-1-ylthio)quinoline-3-carbaldehyde 3
A mixture of 2-mercaptoquinoline-3-carbaldehyde 1 (20 mmol), propargyl bromide 2 (20 mmol), and K2CO3 (22 mmol) in acetone (50 mL) was stirred for 1 h at room temperature. After completion of the reaction (checked by TLC), water was added to mixture of reaction and the precipitated product was filtered off affording 2-(prop-2-yn-1-ylthio)quinoline-3-carbaldehyde 3. This compound was used for the next reaction with no purification.
General procedure for the synthesis of azide derivatives 7a-n
In situ preparation of azide derivatives 7a-n was reported in our pervious works.16
General procedure for the synthesis of quinoline-1,2,3-triazole-acetamide derivatives 8a-n
2-(prop-2-yn-1-ylthio)quinoline-3-carbaldehyde 3 was added to azide derivatives 7a-n to perform a click reaction that used method has already been reported.16
General procedure for the preparation of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n
A mixture of quinoline-1,2,3-triazole-acetamide derivatives 8a-n (1 mmol), hydrazinecarbothioamide 9 (1 mmol), and acetic acid (1 mL) in methanol (5 mL) was heated at reflux for 4 h. After completion of the reaction (checked by TLC), the reaction mixture was cooled down to room temperature and the precipitated product was filtered off and washed by EtOH. The final products were recrystallization in EtOH to give pure quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-phenylacetamide (10a).
Yield: 66%. Light brown powder. Rf value 0.43 (hexane/ethyl acetate = 1/1). M.p.: 186–188 °C. 1H NMR (300 MHz, DMSO-d6) δ 11.82 (s, 1H), 10.45 (s, 1H), 8.92 (s, 1H), 8.51 (s, 1H), 8.43 (s, 1H), 8.19–7.85 (m, 4H), 7.79 (t, J = 7.8 Hz, 1H), 7.57 (d, J = 7.8 Hz, 3H), 7.34 (t, J = 7.7 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 5.30 (s, 2H), 4.74 (s, 2H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 164. 7, 156.5, 147.5, 143.8, 138.9, 137.6, 134.7, 131.4, 129.4, 128.9, 128.0, 126.7, 126.3, 125.9, 125.8, 124.2, 119.7, 52.7, 24.6. Anal. Calcd for C22H20N8OS2: C, 55.45; H, 4.23; N, 23.51; S, 13.45; Found: C, 55.22; H, 4.54; N, 23.77; S, 13.16.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(o-tolyl)acetamide (10b).
Yield: 70%. Light brown powder. Rf value 0.42 (hexane/ethyl acetate = 1/1). M.p.: 191–193 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.55 (s, 1H), 9.78 (s, 1H), 8.92 (s, 1H), 8.50 (s, 1H), 8.42 (s, 1H), 8.02 (dd, J = 32.1, 24.2 Hz, 4H), 7.79 (t, J = 7.8 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.16 (ddd, J = 27.0, 14.3, 7.3 Hz, 3H), 5.34 (s, 2H), 4.73 (s, 2H), 2.20 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 164.8, 156.4, 147.5, 143.8, 137.5, 135.9, 134.7, 132.0, 131.38, 130.9, 128.9, 128.00, 126.7, 126.5, 126.3, 126.0, 125.7, 125.2, 52.4, 24.5, 18.2. Anal. Calcd for C23H22N8OS2: C, 56.31; H, 4.52; N, 22.84; S, 13.07; Found: C, 56.08; H, 4.75; N, 23.08; S, 13.32.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(m-tolyl)acetamide (10c).
Yield: 74%. Light brown powder. Rf value 0.43 (hexane/ethyl acetate = 1/1). M.p.: 193–195 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.82 (s, 1H), 10.37 (s, 1H), 8.92 (s, 1H), 8.51 (s, 1H), 8.42 (s, 1H), 8.18–7.88 (m, 4H), 7.79 (t, J = 7.7 Hz, 1H), 7.59 (t, J = 7.5 Hz, 1H), 7.45–7.28 (m, 2H), 7.21 (t, J = 7.8 Hz, 1H), 6.91 (d, J = 7.5 Hz, 1H), 5.28 (s, 2H), 4.73 (s, 2H), 2.28 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 178.6, 164.6, 156.4, 147.5, 138.8, 138.6, 137.6, 134.8, 131.5, 131.4, 129.2, 128.9, 128.2, 128.0, 126.7, 126.3, 125.9, 124.9, 120.2, 116.9, 52.7, 24.5, 21.6. Anal. Calcd for C23H22N8OS2: C, 56.31; H, 4.52; N, 22.84; S, 13.07; Found: C, 56.12; H, 4.69; N, 22.99; S, 12.91.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(p-tolyl)acetamide (10d).
Yield: 76%. Light brown powder. Rf value 0.42 (hexane/ethyl acetate = 1/1). M.p.: 197–199 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.72 (s, 1H), 10.39 (s, 1H), 8.93 (s, 1H), 8.51 (s, 1H), 8.42 (s, 1H), 8.12 (s, 1H), 8.08–7.91 (m, 3H), 7.85–7.68 (m, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.45 (d, J = 8.1 Hz, 2H), 7.14 (d, J = 8.1 Hz, 2H), 5.27 (s, 2H), 4.73 (s, 2H), 2.27 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 164.4, 156.4, 147.5, 143.7, 137.5, 136.4, 134.7, 133.2, 131.37, 129.7, 128.9, 128.0, 126.7, 126.3, 125.9, 125.8, 119.7, 52.6, 24.5, 20.9. Anal. Calcd for C23H22N8OS2: C, 56.31; H, 4.52; N, 22.84; S, 13.07; Found: C, 56.18; H, 4.76; N, 22.63; S, 12.89.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-ethylphenyl)acetamide (10e).
Yield: 78%. Light brown powder. Rf value 0.40 (hexane/ethyl acetate = 1/1). M.p.: 201–203 °C. IR (KBr, vmax) 3758.61, 2993.23, 2861.35, 2374.77, 2343.23, 1659.27, 1564.65, 1427.01, 1270.36 Cm-1; 1H NMR (301 MHz, DMSO-d6) δ 11.83 (s, 1H), 10.38 (s, 1H), 8.93 (s, 1H), 8.52 (s, 1H), 8.42 (s, 1H), 8.12 (s, 1H), 8.02 (d, J = 8.1 Hz, 2H), 7.94 (dd, J = 8.3, 1.4 Hz, 1H), 7.79 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 7.59 (td, J = 7.4, 7.0, 1.2 Hz, 1H), 7.47 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.3 Hz, 2H), 5.27 (s, 2H), 4.73 (s, 2H), 2.62–2.54 (m, 2H), 1.16 (t, J = 7.6 Hz, 3H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 164.6, 156.4, 147.5, 143.8, 137.5, 135.3, 134.7, 131.4, 128.8, 128.0, 126.8, 126.7, 126.3, 125.9, 125.8, 125.5, 125.4, 52.6, 28.7, 24.5, 16.4. Anal. Calcd for C24H24N8OS2: C, 57.12; H, 4.79; N, 22.21; S, 12.71; Found: C, 56.90; H, 4.98; N, 22.02; S, 12.52.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(2,3-dimethylphenyl)acetamide (10f).
Yield: 72%. Light brown powder. Rf value 0.40 (hexane/ethyl acetate = 1/1). M.p.: 204–206 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.81 (s, 1H), 9.84 (s, 1H), 8.92 (s, 1H), 8.51 (s, 1H), 8.42 (s, 1H), 8.20–7.86 (m, 4H), 7.79 (t, J = 7.7 Hz, 1H), 7.58 (t, J = 7.5 Hz, 1H), 7.23–6.95 (m, 3H), 5.32 (s, 2H), 4.73 (s, 2H), 2.25 (s, 3H), 2.06 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 164.9, 156.4, 147.5, 143.8, 137.6, 137.5, 135.7, 134.7, 131.5, 131.4, 128.9, 128.0, 127.7, 126.7, 126.3, 125.9, 125.7, 125.7, 123.7, 52.4, 24.5, 20.6, 14.4. Anal. Calcd for C24H24N8OS2: C, 57.12; H, 4.79; N, 22.21; S, 12.71; Found: C, 56.87; H, 4.62; N, 22.53; S, 12.96.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(2,6-dimethylphenyl)acetamide (10 g).
Yield: 70%. Light brown powder. Rf value 0.42 (hexane/ethyl acetate = 1/1). M.p.: 210–212 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.81 (s, 1H), 9.84 (s, 1H), 8.92 (s, 1H), 8.55–8.47 (m, 1H), 8.41 (s, 1H), 8.11 (s, 1H), 8.07–7.89 (m, 3H), 7.79 (ddd, J = 8.4, 6.8, 1.5 Hz, 1H), 7.58 (td, J = 7.4, 6.9, 1.2 Hz, 1H), 7.19–7.00 (m, 3H), 5.32 (s, 2H), 4.73 (s, 2H), 2.25 (s, 3H), 2.06 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 164.9, 156.4, 147.5, 143.8, 137.6, 137.5, 135.7, 134.7, 131.5, 131.4, 128.9, 128.0, 127.7, 126.7, 126.3, 125.9, 125.7, 125.7, 123.7, 52.4, 24.5, 20.6, 14.4. Anal. Calcd for C24H24N8OS2: C, 57.12; H, 4.79; N, 22.21; S, 12.71; Found: C, 56.92; H, 4.97; N, 22.43; S, 12.48.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-hydroxyphenyl)acetamide (10 h).
Yield: 68%. Light brown powder. Rf value 0.48 (hexane/ethyl acetate = 1/1). M.p.: 166–168 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.81 (s, 1H), 10.22 (s, 1H), 9.32 (s, 1H), 8.92 (s, 1H), 8.50 (s, 1H), 8.42 (s, 1H), 8.10 (s, 1H), 8.02 (d, J = 8.0 Hz, 2H), 7.94 (d, J = 8.0 Hz, 1H), 7.86–7.72 (m, 1H), 7.66–7.51 (m, 1H), 7.35 (d, J = 8.4 Hz, 2H), 6.72 (d, J = 8.4 Hz, 2H), 5.23 (s, 2H), 4.73 (s, 2H). 13C NMR (76 MHz, DMSO-d6) δ 178.8, 164.2, 156.4, 154.6, 147.5, 143.7, 137.7, 134.7, 131.4, 130.9, 128.8, 128.0, 126.7, 126.3, 125.9, 125.8, 123.1, 116.2, 52.6, 24.6. Anal. Calcd for C22H20N8O2S2: C, 53.64; H, 4.09; N, 22.75; S, 13.02; Found: C, 53.35; H, 3.79; N, 23.01; S, 12.83.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-methoxyphenyl)acetamide (10i).
Yield: 71%. Light brown powder. Rf value 0.44 (hexane/ethyl acetate = 1/1). M.p.: 197–199 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.82 (s, 1H), 10.36 (s, 1H), 8.92 (s, 1H), 8.55–8.46 (m, 1H), 8.42 (s, 1H), 8.12 (s, 1H), 8.05–7.91 (m, 3H), 7.79 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.64–7.54 (m, 1H), 7.52–7.43 (m, 2H), 7.00–6.83 (m, 2H), 5.26 (s, 2H), 4.73 (s, 2H), 3.73 (s, 3H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 164.1, 156.4, 156.0, 147.5, 143.7, 137.6, 134.7, 132.0, 131.4, 128.9, 128.0, 126.7, 126.3, 125.9, 125.8, 121.2, 114.4, 55.6, 52.6, 24.5. Anal. Calcd for C23H22N8O2S2: C, 54.53; H, 4.38; N, 22.12; S, 12.66; Found: C, 56.28; H, 4.64; N, 21.96; S, 12.87.
(E)-N-(4-Bromophenyl)-2-(4-(((3-((2-carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)acetamide (10j).
Yield: 70%. Light brown powder. Rf value 0.45 (hexane/ethyl acetate = 1/1). M.p.: 171–173 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.82 (s, 1H), 10.60 (s, 1H), 8.93 (s, 1H), 8.51 (s, 1H), 8.42 (s, 1H), 8.12 (s, 1H), 8.09–7.99 (m, 2H), 7.94 (d, J = 8.0 Hz, 1H), 7.84–7.75 (m, 1H), 7.63–7.49 (m, 5H), 5.30 (s, 2H), 4.73 (s, 2H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 164.6, 156.4, 147.5, 143.8, 138.0, 137.6, 134.8, 132.5, 131.4, 128.9, 128.0, 126.7, 126.3, 125.9, 125.8, 122.6, 121.5, 52.6, 24.5. Anal. Calcd for C22H19BrN8OS2: C, 47.57; H, 3.45; N, 20.17; S, 11.54; Found: C, 47.27; H, 3.66; N, 19.98; S, 11.28.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(3-nitrophenyl)acetamide (10 k).
Yield: 80%. Brown powder. Rf value 0.48 (hexane/ethyl acetate = 1/1). M.p.: 226–228 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.82 (s, 1H), 10.96 (s, 1H), 8.92 (s, 1H), 8.58 (t, J = 2.2 Hz, 1H), 8.51 (s, 1H), 8.42 (s, 1H), 8.15 (s, 1H), 8.07–7.86 (m, 5H), 7.79 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H), 7.72–7.52 (m, 2H), 5.36 (s, 2H), 4.74 (s, 2H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 165.6, 156.4, 148.4, 147.5, 143.9, 139.9, 137.5, 134.7, 131.4, 130.9, 128.8, 128.0, 126.7, 126.3, 125.9, 125.8, 125.7, 118.8, 113.8, 52.6, 24.5. Anal. Calcd for C22H19N9O3S2: C, 50.66; H, 3.67; N, 24.17; S, 12.29; Found: C, 50.41; H, 3.83; N, 23.98; S, 12.03.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(2-methyl-4-nitrophenyl)acetamide (10 l).
Yield: 84%. Brown powder. Rf value 0.46 (hexane/ethyl acetate = 1/1). M.p.: 218–220 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.82 (s, 1H), 10.01 (s, 1H), 8.92 (s, 1H), 8.51 (s, 1H), 8.42 (s, 1H), 8.21–7.87 (m, 7H), 7.79 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.59 (t, J = 7.5 Hz, 1H), 5.45 (s, 2H), 4.74 (s, 2H), 2.39 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 178.6, 165.8, 156.4, 147.5, 143.0, 142.4, 137.6, 134.7, 131.4, 131.2, 128.1, 128.0, 127.0, 126.7, 126.3, 125.9, 123.7, 122.8, 52.7, 24.2, 18.5. Anal. Calcd for C23H21N9O3S2: C, 51.58; H, 3.95; N, 23.54; S, 11.97; Found: C, 51.33; H, 4.18; N, 23.29; S, 12.21.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-(4-nitrophenyl)acetamide (10 m).
Yield: 75%. Brown powder. Rf value 0.47 (hexane/ethyl acetate = 1/1). M.p.: 185–187 °C. 1H NMR (301 MHz, DMSO-d6) δ 11.83 (s, 1H), 10.20 (s, 1H), 8.92 (s, 1H), 8.52 (s, 1H), 8.42 (s, 1H), 8.25 (d, J = 9.0 Hz, 2H), 8.14 (s, 1H), 8.01 (td, J = 22.4, 20.7, 8.1 Hz, 3H), 7.81 (d, J = 9.2 Hz, 2H), 7.62 (dt, J = 23.9, 7.4 Hz, 2H), 5.39 (s, 2H), 4.74 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 178.7, 164.6, 156.4, 147.5, 145.0, 143.8, 143.0, 137.6, 134.8, 131.4, 128.9, 128.0, 126.7, 126.3, 125.9, 125.8, 124.9, 119.5, 52.8, 24.5. Anal. Calcd for C22H19N9O3S2: C, 50.66; H, 3.67; N, 24.17; S, 12.29; Found: C, 50.38; H, 3.83; N, 24.01; S, 12.51.
(E)-2-(4-(((3-((2-Carbamothioylhydrazineylidene)methyl)quinolin-2-yl)thio)methyl)-1H-1,2,3-triazol-1-yl)-N-phenethylacetamide (10n).
Yield: 75%. Light brown powder. Rf value 0.42 (hexane/ethyl acetate = 1/1). M.p.: 182–184 °C. IR (KBr, vmax) 3758.83, 3053.18, 2820.86, 2375.24, 2342.66, 1665.63, 1543.27, 1489.94, 1308.97 Cm-1;1H NMR (301 MHz, DMSO-d6) δ 11.82 (s, 1H), 8.92 (s, 1H), 8.51 (s, 1H), 8.41 (d, J = 4.5 Hz, 2H), 8.06–7.90 (m, 4H), 7.80 (s, 1H), 7.60 (d, J = 7.6 Hz, 1H), 7.38–7.13 (m, 5H), 5.03 (s, 2H), 4.71 (s, 2H), 3.34–3.26 (m, 2H), 2.72 (t, J = 7.5 Hz, 2H). 13C NMR (76 MHz, DMSO-d6) δ 178.7, 165.7, 156.4, 147.5, 143.7, 139.6, 137.6, 134.7, 131.4, 129.1, 128.8, 128.0, 126.7, 126.6, 126.3, 125.9, 125.5, 123.9, 52.1, 35.3, 24.5. HRMS (ESI) m/z: 504.1072. Anal. Calcd for C24H24N8OS2: C, 57.12; H, 4.79; N, 22.21; S, 12.71; Found: C, 56.86; H, 5.03; N, 22.01; S, 12.50. HPLC: 96.8427%.
In vitro α-glucosidase inhibition and kinetics
The in vitro α-glucosidase inhibitory activity and kinetics of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n were determined according to the previously reported methods.21
Molecular docking and molecular dynamics
The molecular docking of the most potent compounds 10n, 10e, and 10 g and molecular dynamics of the most potent compound 10n in the active site of the modeled α-glucosidase performed using by previously described method.21
Docking study of the most potent compound 10n also was performed on a human α-glucosidase. For this purpose, the pdb structure of 2QMJ (crystal structure of a human α-glucosidase) was taken from the protein data bank (http://www.rcsb.org) as a complex bound with acarbose.22 The active site coordinates of the selected enzyme based on the position of acarbose in the enzyme was identified using by BIOVIA Discovery Studio 3.5, 2019. Center of the grid box was placed at x = − 20.174, y = − 6.206, and z = − 5.281 Å and dimensions of the active site were 50 × 50 × 50 Å. Flexible ligand docking was performed with 100 runs. The best pose of the selected ligands selected for analyzing the interactions. The results were visualized using BIOVIA Discovery Studio 3.5, 2019.
Druglikeness evaluation and ADMET prediction
Druglikeness calculation and ADMET prediction of acarbose and selected compounds 10n, 10e, and 10 g were determined from the preADMET, SwissADMET, and pkCSM online servers.23,24,25
Conclusion
In this study, by hybridizing three known pharmacophores in the potent α-glucosidase inhibitors, a new series of quinoline-thiosemicarbazone-1,2,3-triazole-aceamide derivatives 10a-n were synthesized. These compounds were evaluated against α-glucosidase in vitro and in silico. All synthesized compounds showed significant activity in comparison to positive control acarbose. Lineweaver–Burk plot analysis of most potent compound 10n revealed that this compound was a competitive α-glucosidase inhibitor. Docking and dynamics showed that compound 10n with a favorable BE interacted with important residues in the active site of α-glucosidase. In addition to this compound, other potent compounds, compounds 10e and 10g, also attached to the α-glucosidase active site with more favorable BEs when compared with acarbose. New potent compound 10n, 10e, and 10g has good pharmacokinetic properties and toxicity profile. According to obtained results, here, we presented promising candidates to achieve new drugs with α-glucosidase inhibition mechanism. Although, more preclinical studies are needed to reach this goal.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Roglic, G. WHO Global report on diabetes: A summary. Int. J. Noncommun. Dis. 1, 3–8 (2016).
Dabelea, D. et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. Jama 311, 1778–1786 (2014).
Kulkarni, A., Thool, A. R. & Daigavane, S. Understanding the clinical relationship between diabetic retinopathy, nephropathy, and neuropathy: A comprehensive review. Cureus 16, e56674 (2024).
Kim, Y. M., Jeong, Y. K., Wang, M. H., Lee, W. Y. & Rhee, H. I. Inhibitory effect of pine extract on α-glucosidase activity and postprandial hyperglycemia. Nutrition 21, 756–761 (2005).
Choi, C. W. et al. Yeast α-glucosidase inhibition by isoflavones from plants of Leguminosae as an in vitro alternative to acarbose. J. Agric. Food Chem. 58, 9988–9993 (2010).
Israili, Z. H. Advances in the treatment of type 2 diabetes mellitus. Am. J. Ther. 18, 117–152 (2011).
Baron, A. D. Postprandial hyperglycaemia and α-glucosidase inhibitors. Diabetes Res. Clin. Pract. 40, S51–S55 (1998).
Hossain, U., Das, A. K., Ghosh, S. & Sil, P. C. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food Chem. Toxicol. 145, 111738 (2020).
Viegas-Junior, C., Danuello, A., da Silva Bolzani, V., Barreiro, E. J. & Fraga, C. A. M. Molecular hybridization: a useful tool in the design of new drug prototypes. Curr. Med. Chem. 14, 1829–1852 (2007).
Khalifa, M. M., Sakr, H. M., Ibrahim, A., Mansour, A. M. & Ayyad, R. R. Design and synthesis of new benzylidene-quinazolinone hybrids as potential anti-diabetic agents: In vitro α-glucosidase inhibition, and docking studies. J. Mol. Struct. 1250, 131768 (2022).
Kaur, R., Palta, K. & Kumar, M. Hybrids of isatin-pyrazole as potential α-glucosidase inhibitors: Synthesis, biological evaluations and molecular docking studies. ChemistrySelect 4, 13219–13227 (2019).
Solangi, M. et al. Indole acrylonitriles as potential anti-hyperglycemic agents: Synthesis, α-glucosidase inhibitory activity and molecular docking studies. Bioorg. Med. Chem. 28, 115605 (2020).
Taha, M. et al. Synthesis of quinoline derivatives as diabetic II inhibitors and molecular docking studies. Bioorg. Med. Chem. 27, 4081–4088 (2019).
Moghadam, F. S. et al. Synthesis and structure–activity relationship studies of benzimidazole-thioquinoline derivatives as α-glucosidase inhibitors. Sci. Rep. 13, 4392 (2023).
Wang, G., Peng, Z., Gong, Z. & Li, Y. Synthesis, biological evaluation, and docking studies of novel 5, 6-diaryl-1, 2, 4-triazine thiazole derivatives as a new class of α-glucosidase inhibitors. Bioorg. Chem. 78, 195–200 (2018).
Safapoor, S. et al. Synthesis, ADMT prediction, and in vitro and in silico α-glucosidase inhibition evaluations of new quinoline–quinazolinone–thioacetamides. RSC Adv. 13, 19243–19256 (2023).
Lee, H. W., Yang, J. Y. & Lee, H. S. Quinoline-2-carboxylic acid isolated from Ephedra pachyclada and its structural derivatives show inhibitory effects against α-glucosidase and α-amylase. J. Korean Soc. Appl. Biol. Chem. 57, 441–444 (2014).
Ullah, H. et al. Benzimidazole bearing thiosemicarbazone derivatives act as potent α-amylase and α-glucosidase inhibitors; synthesis, bioactivity screening and molecular docking study. Molecules 27, 6921 (2022).
Wang, G., Peng, Z., Wang, J., Li, X. & Li, J. Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential α-glucosidase inhibitors. Eur. J. Med. Chem. 125, 423–429 (2017).
Emadi, M. et al. Design, synthesis, in vitro anti-α-glucosidase evaluations, and computational studies of new phthalimide-phenoxy-1, 2, 3-triazole-N-phenyl (or benzyl) acetamides as potential anti-diabetic agents. Sci. Rep. 13, 10030 (2023).
Mohammadi-Khanaposhtani, M. et al. New biscoumarin derivatives as potent α-glucosidase inhibitors: Synthesis, biological evaluation, kinetic analysis, and docking study. Polycycl. Aromat. Compd. 40, 915–926 (2018).
Zheng, K. et al. Extraction, identification, and molecular mechanisms of α-glucosidase inhibitory peptides from defatted Antarctic krill (Euphausia superba) powder hydrolysates. Int. J. Biol. Macromol. 266, 131126 (2024).
Seul, S.C. Bioinformatics and Molecular Design Research Center. PreADMET program. (2004).
Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Pires, D. E., Blundell, T. L. & Ascher, D. B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 58, 4066–4072 (2015).
Asgari, M. S. et al. Biscoumarin-1, 2, 3-triazole hybrids as novel anti-diabetic agents: Design, synthesis, in vitro α-glucosidase inhibition, kinetic, and docking studies. Bioorg. Chem. 92, 103206 (2019).
Acknowledgements
We thankfully acknowledge the support of Biomedical and Microbial Advanced Technologies (BMAT) Research Center of Babol University of Medical Sciences (Project code: 724135069 and ethical code: IR.MUBABOL.HRI.REC.1402.037).
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
M.M–K., R.A., M.H. and M.M. designed the research work, performed the docking study, and wrote the manuscript. A.H.A., M.N., and N.D. synthesized and purified the compounds, and carried out 1H NMR and 13C NMR. M.A. supervised the biological tests. A.K., S.M. and M.A.F performed the biological tests. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khademian, A., Halimi, M., Azarbad, R. et al. Quinoline-thiosemicarbazone-1,2,3-triazole-acetamide derivatives as new potent α-glucosidase inhibitors. Sci Rep 14, 30876 (2024). https://doi.org/10.1038/s41598-024-81668-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-81668-5
