
Abstract
Recent studies have found that disulfidptosis occurs in cells under glucose starvation. The role of this programmed death method in gastric cancer remains to be explored. Cluster analysis based on disulfidptosis related genes to analyze the differential characteristics of disulfidptosis subtypes. We construct a prognostic risk model using 12 differentially expressed genes of disulfidptosis subtypes. We also analyzed the disulfidptosis subtypes at single-cell resolution. We found that cluster 1 has a poor prognosis and is characterized by a younger age. Inhibiting the expression of GAMT genes associated with disulfidptosis subtypes can significantly inhibit the proliferation of gastric cancer cells, which may be an important target for gastric cancer treatment. Cluster 2 patients are more sensitive to various chemotherapy drugs and immunotherapy. Mesenchymal cells, especially myCAF, endothelial cells, and smooth muscle cells, have strong disulfidptosis scores. In summary, our study provides new insights into the role of disulfidptosis in gastric cancer, and this may be used to guide the treatment of gastric cancer.
Similar content being viewed by others


Introduction
Gastric cancer (GC) ranks as the sixth most prevalent malignancy and stands as the third leading cause of cancer-related fatalities globally1. Presently, early detection of GC is contingent upon gastroscopy; nevertheless, this diagnostic procedure is not conducted annually in numerous regions, leading to delayed diagnoses for numerous GC patients, often at advanced stages. The therapeutic approaches for GC have traditionally hinged on clinicopathological evaluations of tumors, encompassing surgical interventions, diverse chemotherapy regimens, and immunotherapies targeting immune checkpoints. Nonetheless, owing to the inherent heterogeneity of GC, these interventions exhibit efficacy only in select patients. Hence, imperative is the need to categorize tumors based on their intrinsic heterogeneity, delineate their correlation with tumor response to treatment, and refine treatment strategies accordingly.
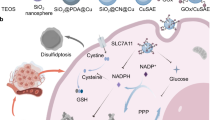
Regulated cell death is a form of cell death that can be modulated through specific molecular pathways or via genetic and pharmacological interventions. Recent studies have identified a metabolism-related form of programmed cell death known as disulfidptosis. This form of cell death was observed in renal cancer cells where high expression of SLC7A11 accelerates cytosolic NADPH depletion under glucose starvation2. This depletion prevents the reduction of disulfide bonds, leading to disulfide bond stress and ultimately triggering disulfidptosis. Disulfide protein denaturation is a new cell death pathway distinct from other programmed cell death mechanisms. This stress, caused by abnormal disulfide accumulation, exerts highly toxic effects on cells, affecting the survival and proliferation of cancer cells.
Disulfide metabolism also influences bioactivity related to cancer cells2. Liu and colleagues further explored the anticancer effects of GLUT-targeting inhibitors, finding that these inhibitors significantly suppressed tumor growth both in vivo and in vitro3. Therefore, disulfidptosis has the potential to become an emerging field in cancer therapy. However, comprehensive research and exploration are essential to clarify its precise mechanisms and therapeutic applications.
Here, we divided gastric cancer into two subgroups based on the expression data of disulfidptosis-related genes. We explored the differences between the subtypes from various perspectives. Using the differential genes between subtypes, we established a prognostically excellent risk model. Additionally, we analyzed the treatment preferences of the subtypes and found that subtype 2 seems to benefit more from immune checkpoint inhibitors and various chemotherapies. Finally, we delved into the differential infiltration of tumor epithelial cells and stromal cells in different subtypes from a single-cell perspective.
Materials and methods
Data acquisition and processing
Gene expression profiles and clinical information for the TCGA-STAD cohort sourced from the Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) were acquired using the TCGAbiolinks package within the R programming environment4. Conversion of the count values for TCGA-STAD cohort samples to transcripts per million (TPM) was carried out. Additionally, somatic mutations and copy number variants within the TCGA-STAD cohort were retrieved utilizing the TCGAbiolinks package in R. The analysis of somatic mutation data was further conducted using the R package maftools5.
Multiple data (GSE622546, GSE154597, GSE269428, GSE844379, GSE5730310 and GSE3494211) were downloaded from the Gene-Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/gds/). Because of the large number of datasets on the GPL570 platform, four datasets (GSE62254, GSE15459, GSE57303 and GSE34942) from the GPL570 platform were merged as one dataset named as GPL570 meta-dataset using “oligo” package in R12. The gastric cancer immunotherapy cohort (PRJEB25780) was downloaded from European Nucleotide Archive (ENA, https://www.ebi.ac.uk)13.
We analyzed the largest single-cell sequencing dataset in gastric cancer (GSE183904)14 and applied the following quality control criteria: genes had to be expressed in more than three cells, and only cells expressing 3,000–6,000 genes were included. Cells with mitochondrial RNA > 20% were excluded, and doublets were removed using the DoubletFinder package15.
Construction of the disulfidptosis metabolism-related clusters
We gathered 24 disulfidptosis related genes and performed unsupervised clustering on TCGA-STAD dataset using the ConsensusClusterPlus package based on the expression data of these genes16. To apply the subtypes established in the TCGA-STAD dataset to other datasets, we first calculated the average expression of all cells in each sample for the single-cell dataset. Subsequently, we employed the Support Vector Machine (SVM) algorithm to train a classification model on TCGA-STAD, and then classified the samples in the GSE183904 dataset into two subtypes. For regular bulk sequencing datasets, we directly utilized standardized expression data.
Pathway enrichment analyses
In order to explore the differences in signaling pathways between subtypes, we utilized the Gene Set Enrichment Analysis (GSEA) software, using KEGG pathways as gene sets to analyze the differential enrichment of pathways between subtypes. NOM p-value < 0.05 were considered statistically significant. Gene Set Variation Analysis (GSVA) was employed to assess the enrichment levels of signaling pathways in individual samples.
The disulfidptosis subtype characteristic score construction
Due to significant prognostic differences between the two disulfidptosis subtypes, we aimed to establish a prognostic risk assessment model based on differential features between these two subtypes. Initially, we analyzed differentially expressed genes between the two subtypes, with the filtering criteria set at a false discovery rate (FDR) < 0.05 and |log2 fold change (FC)| > 1.0. To enhance the performance of our model, we also selected genes significantly associated with prognosis in univariate Cox regression analysis (p < 0.01). Subsequently, the LASSO-Cox algorithm was employed to choose the optimal model genes in TCGA-STAD dataset and establish the prognostic model. This prognostic model was then validated in other datasets (GPL570, GSE26942, GSE84437). As the TCGA-STAD data were based on RNA-seq technology, while the two validation datasets were derived from microarray data, all expression data were log10-transformed and then Z-score normalized before training and validating the model.
Cell culture and the construction of GAMT knockout cell lines
Human gastric cancer cell lines (AGS) were obtained from the Cell Bank of the Typical Culture Preservation Committee of the Chinese Academy of Sciences (Shanghai, China). All cells were cultured in RPMI-1640 medium (GIBCO, Grand Island, USA) supplemented with 10% fetal bovine serum (FBS) (BI, USA) and a commercial antibiotic cocktail of penicillin and streptomycin (Procell, China) at 37 °C with 5% CO2 in a fully humidified environment.
The lentivirus carrying GAMT sgRNA and control sgRNA was purchased from Tongyong (Tongyong Biotechnology Co. Ltd. Anhui, China). Knockout cell lines were generated by lentivirus infection and puromycin selection according to the manufacturer’s instructions. The target sequences of GAMT can be found in Supplementary Table 1.
Quantitative real-time PCR
Total RNA was extracted from cell samples using TRIzol (Thermofisher, USA). GAPDH (Glyceraldehyde-3-phosphate dehydrogenase) was chosen as the internal reference gene for normalization. The 2^-ΔΔCt method was employed to calculate the relative expression levels of the target genes. Statistical analysis was conducted using GraphPad Prism software, with differences between groups evaluated by Student’s t-test, where a p-value less than 0.05 was considered statistically significant. mRNA primer sequences can be found in Supplementary Table 1.
Transwell assay, CCK8 assay and EDU assay
The Transwell experiment was conducted in a 24 well plate, with the upper chamber containing 200 µ L serum-free medium, filled with 550 in the lower chamber µ L culture medium containing 10% FBS. AGS was inoculated at a density of 20,000 cells per well. We fix the cells at 48 h.
The cells were seeded into 96-well plates at a density of 3000 cells/well for AGS cells. At 0, 24, 48, and 72 h, 10 µL of CCK8 reagent was added to each well, followed by incubation at 37 °C for 2 h. The absorbance at 450 nm was measured.
5-ethynyl-2’-deoxyuridine (EdU) incorporation assay using the EdU Cell Proliferation Kit with Alexa Fluor 647 (Beyotime, Shanghai, CHN) was performed to examine the cell proliferation capacity. The cells were seeded into 96-well plates at a density of 12,000 cells/well for AGS cells. We detect these cells after 24 h.
Western blotting
Cell lysates were extracted using RIPA buffer supplemented with protease inhibitors. Protein concentrations were determined using a BCA Protein Assay Kit (Cwbio Biotechnology, China). Equal amounts of protein (20 µg) were resolved by SDS-PAGE and transferred onto PVDF membranes. The membranes were blocked with 5% BSA in TBST for 1 h at room temperature, followed by overnight incubation at 4 °C with primary antibodies. After three 10-minute washes with TBST, the membranes were incubated with secondary antibodies diluted in blocking buffer for 1 h at room temperature. Subsequently, the membranes were washed three more times with TBST, and protein bands were visualized using an enhanced chemiluminescence (ECL) detection kit (Yeasen Biotechnology, Shanghai, China). Quantification of band intensity was performed using ImageJ software. GAMT antibody (mouse monoclonal, Cat#66322-1-Ig, used at 1:2000) and β-actin antibody (recombinant, Cat#81115-1-RR, used at 1:20000) was purchased from Proteintech (Wuhan, China).
Additional bioinformatic and statistical analyses
We inferred single-cell compositions and their contributions to global gene expression changes from bulk RNA sequencing by BayesPrismm package17. All of the above analyses were performed using the R software (version 4.0.2, http://www.rproject.org). Statistical differences not specifically stated were set as P < 0.05.
Result
Identification of disulfidptosis subtypes and in gastric cancer
To investigate the expression characteristics of disulfidptosis-related genes in gastric cancer (GC), we performed clustering analysis on TCGA-STAD cohort samples, focusing on 24 disulfidptosis-related genes. This analysis revealed two distinct subtypes (Fig. 1A and B). In Kaplan-Meier survival curves, patients in cluster 2 (C2) exhibited a better survival outcome compared to those in cluster 1 (C1) (Fig. 1C). Interestingly, we observed that the proportion of patients in C1 seemed to be significantly higher among those under 50 years old, while relatively fewer patients were in the 71–80 age group (Fig. 1D). Previous studies identified four subtypes of gastric cancer in the TCGA-STAD dataset18. In our analysis, significant differences emerged in the distribution of C1 and C2 among the MSI and GS subtypes in the TCGA dataset (Fig. 1D).

Identification of GC disulfidptosis-related subtypes. (A and B) Heatmap displaying consensus clustering with the robust classification (k = 2). (C) Kaplan-Meier curves of OS between clusters in the TCGA-STAD cohort. Log-rank test p values are shown. (D) Bar plots showing the distribution of TCGA subtypes, Lauren subtypes and clinical traits among the metabolic subtypes.
Disulfidptosis subtypes show distinct signaling pathway features
Ten tumor-related signaling pathways also exhibit distinct patterns between the two subtypes. C2 showed high enrichment of MYC, cell cycle, NRF2, and P53 pathways (Fig. 2A). Analysis of hallmark pathways revealed significant enrichment of the hypoxia and angiogenesis pathway in C1, suggesting a prominent hypoxic feature in this subtype (Fig. 2B). This raises the question of whether disulfidptosis is associated with hypoxia. In contrast, C2 displayed greater enrichment of metabolism-related pathways, such as MTORC1 signaling, fatty acid metabolism, oxidative phosphorylation and glycolysis (Fig. 2B). Similar results were obtained through GSEA (Fig. 2C).

Signaling pathway heterogeneity among two disulfidptosis-related subtypes. (A) Comparison of 10 cancer-related pathways among two disulfidptosis-related subtypes. (B) Comparison of hallmark pathways among two disulfidptosis-related subtypes. (C) KEGG signaling pathway in disulfidptosis-related subtypes using Gene set enrichment analysis.
Gene mutation of GC subtypes
Genomic instability (GI) plays a crucial role in the initiation and progression of tumors. We found that tumor mutation burden (TMB) is significantly higher in C2 compared to C1 (Fig. 3A). To further investigate, we analyzed gene mutations differences between the subtypes. The waterfall plot reveals that among the top 20 mutated genes in gastric cancer, seven exhibit significant differential mutations between C1 and C2 (Fig. 3B and C). Notably, TTN and TP53, which have the highest mutation rates in gastric cancer, show higher mutation rates in C2, aligning with the higher TMB in this subtype.

(A) Comparisons of TMB scores between two disulfidptosis-related subtypes. (B) Top 20 mutated genes in all gastric cancer patients of TCGA-STAD cohort. (C) Top 20 differentially mutated genes between disulfidptosis-related subtypes in TCGA-STAD.
Construction and validation of a prognostic model for disulfidptosis
Due to significant prognostic differences between the two disulfidptosis subtypes, we aimed to establish a prognostic risk assessment model based on differential features between C1 and C2. We identified 158 differentially expressed genes. To mitigate the impact of sequencing technology differences, we initially performed log10 transformation on all data and subsequently applied z-score normalization before analysis. The LASSO-Cox algorithm was employed to identify the most robust prognostic model for disulfidptosis. Finally, risk models associated with cancer functional status pathways were developed as follows: 0.0364*APOD + 0.0040*ARL14EPL + .0489*GAMT + .0370*GDF6 + .0991*KCND2 + .01333*KRT82 + .02449*NGF + .0043*PLCL1 + .1095*PPDPFL + .0488*PRAC1 + .01013*PRICKLE1 + .0031*SYT6 (Fig. 4A). Kaplan-Meier curves indicated that our risk scoring model effectively distinguishes prognoses (Fig. 4B), and its performance was validated in independent datasets (Fig. 4C and E).

(A) Partial likelihood deviance for the lasso regression and Lasso regression analysis. (B) Patients were divided into high-risk and low-risk subgroup based optimal cut-off, Kaplan-Meier analysis demonstrated that patients with higher risk score exhibited worse overall survival in TCGA-STAD. (C-E) The prognostic difference was validated in GPL570 cohort, GSE26942 and GSE84437. (F) qPCR analysis of GAMT expression in indicated AGS cells (n = 3 well/group, one-way ANOVA with Tukey’s multiple comparisons test on all pairwise combinations). (G) Western blotting analysis of GAMT expression in indicated AGS cells. original blots/gels are presented in Supplementary Figure S1. (H) The cell viability of AGS cells was significantly inhibited by the CCK- 8 assay (At 0, 24, 48, and 72 h). (I) Compared with the control group, the proliferation rate of AGS cells was significantly inhibited after GAMT knockout by EdU staining (Detect these cells at 24 h.). (J) Transwell experiments showed that the migratory ability of AGS cells was inhibited after GAMT knockout (Fix the cells at 48 h.).
To explore the role of model genes in GC, we chose the PPDPFL gene with the highest model coefficient, but we found that the expression level of the PPDPFL gene was too low in GC cell lines, which may account for its high model coefficient. After our screening, we decided to study the role of GAMT in GC. We found that the expression of the GAMT gene was significantly suppressed (Fig. 4F and G). Knockdown experiments with GAMT-sg1 demonstrated that inhibiting GAMT significantly reduced the proliferation ability in vitro (Fig. 4H and I). Transwell assays showed that GAMT knockout suppressed the migration of AGS cells (Fig. 4J). Knockdown of GAMT leads to decreased proliferation and migration, which is reversed after re-expression of GAMT cDNA (Fig. 4H, I and J).
Disulfidptosis subtype-associated treatment strategy for GC
To predict chemotherapy responses, we utilized the Cancer Genome Project (CGP) database, identifying significant differences in estimated IC50 values for five drugs between the two subtypes (Fig. 5A and E). Notably, patients in the C2 subtype exhibited greater sensitivity to treatment for all these drugs, which may explain the better prognosis in this group. We found that after GAMT knockout, the IC50 of these drugs significantly decreased in AGS cells, indicating that GAMT knockout enhances the sensitivity of gastric cancer cells to these chemotherapeutic agents (Figure F-J).

(A–E) The chemotherapy response of two subtypes for five common chemotherapy drugs (5-Fluorouracil, oxaliplatin, docetaxel, mitomycin C and paclitaxel). (F–J) The IC50 values for five common chemotherapy drugs in AGS cells transfected with indicated lentivirus (Nonlinear regression analysis ;5-Fluorouracil, oxaliplatin, docetaxel, mitomycin C and paclitaxel). (K and L) Proportion of patients with response to immunotherapy in two disulfidptosis-related subtypes in the PRJEB25780 cohort. CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease. CR/PR was identified as responder, and SD/PD was identified as non-responder.
The subtype data from TCGA-STAD were utilized to train a model using the Support Vector Machine (SVM) algorithm, and subsequently, the trained model was employed to predict subtypes in the PRJEB25780 dataset. Next, we investigated whether there were differences in immune therapy responses between patients in the two subtypes. Interestingly, we observed that the C2 subtype appeared to be more sensitive to immune therapy (Fig. 5F and G). This finding might potentially guide clinical treatment strategies.
Dissection of tumor microenvironment based on single cell transcriptome
To investigate the differences in the tumor microenvironment between the two disulfidptosis subtypes, we analyzed single-cell mRNA expression profiles from 26 gastric cancer tissues in the GSE183904 dataset. After quality control, we classified the filtered cells into eight major cell types using classical biomarkers (Supplementary Table 2). We calculated the average expression of individual genes for all cells in each single-cell sample and considered this average as the expression at the bulk level for each single-cell sample. Subsequently, using SVM algorithm, we trained a subtype model on the TCGA-STAD dataset and successfully classified GSE183904 into two disulfidptosis subtypes (Supplementary Table 3). We employed the AUCell algorithm to calculate the scores for the gene set of 24 disulfidptosis-related genes in each cell. We observed higher disulfidptosis scores in stromal cells and tumor epithelial cells, while immune cells exhibited lower scores (Fig. 6A). In the TCGA-STAD dataset, the distribution proportions of the two inflammatory cancer-associated fibroblasts (iCAFs) cell clusters between the two subtypes did not seem significantly different (Fig. 6B and C). Simultaneously, the disulfidptosis scores in these two iCAF cell subtypes did not exhibit significant differences between the two cell subtypes (Fig. 6D and E). Conversely, myo-cancer associated fibroblasts (myCAFs), endothelial cells, and smooth muscle cells had higher proportions in C1 (Fig. 6F and H). Interestingly, disulfidptosis scores for myCAFs were higher in C2, while those for endothelial and smooth muscle cells were higher in C1(Fig. 6I and K). It is noteworthy that TCGA-STAD data suggested that patients with higher proportions of myCAF, endothelial cells, and smooth muscle cells had poorer prognoses, indicating that these three cell types may promote tumor progression (Fig. 6L and N). Conversely, the C2 subtype had a higher proportion of tumor_1 cells, which were linked to better prognosis (Fig. 6O and P). We analyzed the characteristics of tumor cell and found that, compared to tumor_2, tumor_1 had a lower degree of malignancy (Fig. 6Q and S), further supporting the better prognosis in patients with higher tumor_1 proportions.

Dissection of tumor microenvironment based on single cell transcriptome. (A) Comparisons of disulfidptosis score among two disulfidptosis-related subtypes within all cell clusters. (B and C, F–H, O) Boxplots show the distribution of proportion of iCAF_1, iCAF_2, myCAF, endothelial cells, smooth muscle cells and tumor_1 among two disulfidptosis-related subtypes in TCGA-STAD. (D and E, I–K) Boxplots show comparisons of disulfidptosis score among two disulfidptosis-related subtypes. (L–N, P) Kaplan-Meier analysis of high- and low cell infiltration in TCGA-STAD. (Q–S) Comparing CEACAM5, CEACAM6 and KRAS expression across two malignant epithelial cell clusters.
Discussion
GC remains a major challenge in personalized medicine, particularly due to the resistance of tumors to cell death and the influence of metabolic disadvantages on cancer cell activity. Disulfidptosis, a novel form of cell death, has shown potential as a therapeutic target in cancer treatment. In our study, we classified GC into two distinct subtypes based on disulfidptosis-related gene expression. Using these subtype-specific genes, we developed a robust prognostic risk model, which demonstrated good predictive performance and was further validated on independent datasets.
Our findings revealed that subtype C2 exhibits higher sensitivity to immune checkpoint inhibitors and various chemotherapy agents, suggesting that patients in this group may respond more favorably to these treatments. Through single-cell analyses, we also observed distinct patterns in the infiltration of tumor and stromal cells between the two subtypes, which offers insights into their differential responses to treatment. Consistent with previous studies linking metabolic stress with therapeutic response, we found that subtype-specific disulfidptosis gene expression may influence clinical outcomes in GC, warranting further exploration of the metabolic vulnerabilities in subtype C1.
In the TCGA-STAD cohort, we identified that C1 patients, who have a poorer prognosis, are more often under 50 years old. Hypoxia and angiogenesis pathways were enriched in C1, aligning with literature on their role in promoting tumor malignancy and potentially contributing to resistance to disulfidptosis. This raises an interesting hypothesis that hypoxia could confer resistance to disulfidptosis, thus impacting cell death in GC. Future research is needed to clarify these mechanisms and explore hypoxia as a modulator of disulfidptosis resistance.
Our analysis identified GAMT as a gene of interest, with functional studies showing that silencing GAMT inhibits GC cell proliferation and invasion. This suggests that targeting GAMT may offer a promising therapeutic avenue for GC treatment. Given that cells with high SLC7A11 expression are more susceptible to disulfide death during glucose starvation, combining GLUT inhibitors with GAMT-targeted therapies could enhance treatment efficacy in patients with high SLC7A11 expression, a hypothesis we plan to explore further.
AS GC patients show variable sensitivity to chemotherapy and immunotherapy, we observed that C2 subtype patients were more responsive to these treatments, including immunotherapy, in an independent GC dataset. This suggests that disulfidptosis-based subtyping could inform personalized treatment regimens, allowing clinicians to tailor therapies according to the molecular profile of each patient’s tumor.
Our single-cell analysis uncovered that myCAF cells exhibit an inverse disulfidptosis score compared to endothelial and smooth muscle cells, despite all three cell types being implicated in tumor progression. This points to potentially diverse pathways through which disulfidptosis impacts different cell types within the tumor microenvironment, an area that warrants further investigation to clarify these cellular interactions and inform targeted therapies.
Conclusion
In summary, while our study highlights the prognostic and therapeutic relevance of disulfidptosis-related subtypes in GC, continued research is essential to fully understand the mechanistic pathways involved and optimize the clinical application of our findings.
Data availability
The data that support the findings of this study are available in GEO (https://www.ncbi.nlm.nih.gov/geo/, GSE62254, GSE15459, GSE57303, GSE34942, GSE26942, GSE84437 and GSE183904), TCGA (https://portal.gdc.cancer.gov/repository, TCGA-STAD and other tumor cohorts), and the Supporting Information. Then, raw transcriptome and clinical data of immunotherapy cohort (PRJEB25780) were retrieved from European Nucleotide Archive.
Abbreviations
- GC:
-
Gastric cancer
- TCGA:
-
The Cancer Genome Atlas
- TPM:
-
Transcripts per million
- GEO:
-
Gene-Expression Omnibus
- ENA:
-
European Nucleotide Archive
- SVM:
-
Support Vector Machine
- GSEA:
-
Gene Set Enrichment Analysis
- GSVA:
-
Gene Set Variation Analysis
- FDR:
-
False discovery rate
- C1:
-
Cluster 1
- C2:
-
Cluster 2
- GI:
-
Genomic instability
- TMB:
-
Tumor mutation burden
- iCAFs:
-
Inflammatory cancer-associated fibroblasts
- myCAFs:
-
Myo-cancer associated fibroblasts
References
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 Countries. CA Cancer J. Clin. 71(3), 209–249 (2021).
Liu, X. et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 22(4), 476–486 (2020).
Liu, X. et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat. Cell. Biol. 25(3), 404–414 (2023).
Colaprico, A. et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44 (8), e71 (2016).
Mayakonda, A., Lin, D. C., Assenov, Y., Plass, C. & Koeffler, H. P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 28(11), 1747–1756 (2018).
Cristescu, R. et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 21(5), 449–456 (2015).
Ooi, C. H. et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. PLoS Genet. 5(10), e1000676 (2009).
Oh, S. C. et al. Clinical and genomic landscape of gastric cancer with a mesenchymal phenotype. Nat. Commun. 9(1), 1777 (2018).
Yoon, S. J. et al. Deconvolution of diffuse gastric cancer and the suppression of CD34 on the BALB/c nude mice model. BMC cancer 20(1), 314 (2020).
Qian, Z. et al. Whole genome gene copy number profiling of gastric cancer identifies PAK1 and KRAS gene amplification as therapy targets. Genes Chromosom. Cancer 53(11), 883–894 (2014).
Chia, N. Y. et al. Regulatory crosstalk between lineage-survival oncogenes KLF5, GATA4 and GATA6 cooperatively promotes gastric cancer development. Gut 64(5), 707–719 (2015).
Carvalho, B. S. & Irizarry, R. A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 26(19), 2363–2367 (2010).
Kim, S. T. et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 24(9), 1449–1458 (2018).
Kumar, V. et al. Single-cell atlas of lineage states, tumor microenvironment, and subtype-specific expression programs in gastric cancer. Cancer Discov 12(3), 670–691 (2022).
McGinnis, C. S., Murrow, L. M., Gartner, Z. J. & Doubletfinder doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell. Syst. 8(4), 329–337e324 (2019).
Wilkerson, M. D. & Hayes, D. N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinf. (Oxford England). 26(12), 1572–1573 (2010).
Chu, T., Wang, Z., Pe’er, D. & Danko, C. G. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. cancer. 3(4), 505–517 (2022).
Comprehensive molecular characterization. of gastric adenocarcinoma. Nature 513(7517), 202–209 (2014).
Acknowledgements
Not Applicable.
Funding
None
Author information
Authors and Affiliations
Contributions
Conception and design, Qi Zhou and Wenxue Wu; manuscript writing, Qi Zhou, Wenxue Wu, Guoqiang Tao and Zhiqing Chen; acquisition of data, Guoqiang Tao and Zhiqing Chen; analysis and interpretation of data, Qi Zhou, Wenxue Wu, Guoqiang Tao and Zhiqing Chen. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Wenxue Wu, Guoqiang Tao and Zhiqing Chen have contributed equally to this work and share first authorship.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, W., Tao, G., Chen, Z. et al. Identification of disulfidptosis-related subtypes in gastric cancer and GAMT is a key gene during disulfidptosis. Sci Rep 15, 111 (2025). https://doi.org/10.1038/s41598-024-83580-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-83580-4
