
Abstract
Diabetes mellitus, particularly type 2 diabetes, is a growing global health challenge characterized by chronic hyperglycemia due to insulin resistance. One therapeutic approach to managing this condition is the inhibition of α-glucosidase, an enzyme involved in carbohydrate digestion, to reduce postprandial blood glucose levels. In this study, a series of thiosemicarbazide-linked quinoline-piperazine derivatives were synthesized and evaluated for their α-glucosidase inhibitory activity, to identify new agents for type 2 diabetes management. Structure-activity relationship (SAR) analysis revealed that the nature and position of substituents on the aryl ring significantly impacted the inhibitory potency. Among the synthesized derivatives, the 2,5-dimethoxy phenyl substitution (7j) exhibited the most potent activity with an IC50 value of 50.0 µM, demonstrating a 15-fold improvement compared to the standard drug acarbose. Kinetic studies identified compound 7j as a competitive inhibitor, with a Ki value of 32 µM. Molecular docking simulations demonstrated key interactions between compound 7j and the active site of α-glucosidase, while molecular dynamics simulations confirmed the stability of the enzyme-ligand complex, reflected in low RMSD and RMSF values.
Similar content being viewed by others
Introduction
Diabetes mellitus is a chronic metabolic disorder characterized by elevated blood glucose levels (hyperglycemia) due to disruption in insulin secretion, insulin action, or both. The disease affects millions of people worldwide and is associated with various complications, including cardiovascular diseases, kidney failure, neuropathy, and retinopathy1. Diabetes is typically classified into two major types: type 1 diabetes, resulting from autoimmune destruction of pancreatic β-cells, and type 2 diabetes, which is associated with insulin resistance and inadequate insulin production2. Type 2 diabetes accounts for approximately 90–95% of all diabetes cases and is closely linked to lifestyle factors such as diet and physical activity3.
One of the primary goals of diabetes management is to regulate blood sugar levels and prevent postprandial hyperglycemia, which is a major contributor to the long-term complications of diabetes4. Among the therapeutic strategies employed to manage postprandial glucose levels, α-glucosidase inhibitors play a significant role. α-Glucosidase is an enzyme located in the brush border of the small intestine that plays a key role in carbohydrate digestion. It catalyzes the breakdown of complex carbohydrates, such as starch and disaccharides, into simple sugars like glucose5. The inhibition of α-glucosidase slows down the digestion and absorption of carbohydrates, thereby reducing the rapid rise in blood glucose levels after a meal6.
α-Glucosidase inhibitors are an essential class of drugs for managing type 2 diabetes. By delaying carbohydrate digestion, these inhibitors prevent sudden increases in blood glucose, helping to maintain better glycemic control. Common α-glucosidase inhibitors, such as acarbose, miglitol, and voglibose, are widely used in clinical practice7. Developing novel α-glucosidase inhibitors with improved efficacy and reduced side effects is an ongoing area of research in medicinal chemistry. Through structure-activity relationship (SAR) studies, researchers aim to discover new inhibitors that offer better therapeutic outcomes and minimal adverse effects, such as gastrointestinal discomfort commonly associated with current inhibitors8.
Recent advances in the design of α-glucosidase inhibitors have focused on exploring a variety of heterocyclic scaffolds, including quinoline9, isatin10, and benzimidazole11,12 derivatives. Among these, quinoline-based α-glucosidase inhibitors have garnered significant attention due to their promising bioactivity and structural versatility. Numerous series of α-glucosidase inhibitors containing a quinoline ring have been synthesized, with structural modifications aimed at enhancing potency and specificity13,14,15. For example, quinoline carboxylic acid has demonstrated significant inhibitory potency compared to the standard acarbose (IC50 = 66.5 ± 1.5 µg/mL)16. Additionally, Schiff bases derived from quinoline analogs (Compound A, Fig. 1) have shown IC50 values ranging from 6.20 to 48.50 µM. SAR studies revealed that the presence of electron-donating groups in these analogs generally diminished inhibitory activity, suggesting that electron-withdrawing substituents may enhance binding interactions with the enzyme’s active site17. Compound B also exhibited highly potent α-glucosidase inhibitors.
Thiosemicarbazide moieties have also emerged as potent α-glucosidase inhibitors. For instance, thiosemicarbazide-isatin derivatives (C) exhibit impressive inhibitory activity, with IC50 values ranging between 1.20 ± 0.10 and 35.60 ± 0.80 µM, significantly outperforming acarbose (IC50 = 38.60 µM)18. Also in another study, chromones conjugated to thiosemicarbazide (D, IC50 = 0.11 ± 0.01 µM), demonstrated superior activity, suggesting that the thiosemicarbazide framework provides a strong basis for further development of α-glucosidase inhibitors19.
Similarly, aryl-piperazine-linked benzimidazole derivatives have shown noteworthy inhibitory activity against α-glucosidase. The most potent analog, compound E, acts as a competitive inhibitor with a Ki value of 120 µM. Molecular docking studies reveal key interactions with amino acid residues contributing to the inhibitory effect20. The benzimidazole-piperazine derivatives (compound F) exhibited significant inhibition against both α-glucosidase (IC50 = 0.85–29.72 µM) and α-amylase (IC50 = 4.75 − 40.24 µM), further highlighting their therapeutic potential21. A very recent study applied a fused design strategy by incorporating a quinoline core with a flexible piperazine linker (compound G), yielding promising results22.
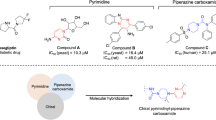
In the current study, fusing and fragment-based design strategies were employed by combining distinct pharmacophores—thiosemicarbazide, quinoline, and piperazine—into a single molecule to achieve enhanced therapeutic effects. The newly designed compounds were evaluated for their α-glucosidase inhibitory activity, with their inhibition mechanisms further investigated through kinetic studies, molecular docking, and molecular dynamics simulations. These computational analyses provided a comprehensive understanding of the molecular interactions between the synthesized compounds and the enzyme, highlighting key structural features that contribute to their inhibitory activity. These findings offer a robust foundation for future optimization and the development of more potent α-glucosidase inhibitors.

The rationale for the design of substituted piperazine conjugated to quinoline-thiosemicarbazide as new α-glucosidase inhibitors
Results and discussion
Chemistry
In this work, we synthesized a novel series of 2-(4-(3-((2-carbamothioylhydrazineylidene)methyl)quinolin-2-yl)piperazin-1-yl)-N-phenylacetamide derivatives 7a–n by reaction 2-(4-(3-formylquinolin-2-yl)piperazin-1-yl)-N-phenylacetamide 6a-n and thiosemicarbazide. In the previous step, nucleophilic reaction between 2-(piperazin-1-yl)quinoline-3-carbaldehyde 4 and 2-chloro-N-phenylacetamide derivatives 5a-n in the presence of K2CO3 leads to synthesis of 2-(4-(3-formylquinolin-2-yl)piperazin-1-yl)-N-phenylacetamide 6a-n derivatives. At this step, substances 4 and 1 eq K2CO3 were initially mixed in DMF for half an hour at room temperature. Then, 2-chloro-N-phenylacetamide was added to the reaction mixture, and temperature was raised to 80 °C. After completing the reaction, water was added to the reaction mixture, resulting in the formation of a yellow precipitate. The precipitate was filtered and washed several times with water. As outlined in Scheme 1, via Vilsmeier–Haack process chloroquinoline-3-carbaldehyde 2 was synthesized then reaction between compound 2 and 1-Boc-piperazine in the presence of KI and K2CO3 and deprotection of tert-butyl 4-(3-formylquinolin-2-yl)piperazine-1-carboxylate 3 leads to the synthesis of 2-(piperazin-1-yl)quinoline-3-carbaldehyde 4 (Scheme 1).

Synthesis of 2-(4-(3-(1 H-benzo[d]imidazol-2-yl)quinolin-2-yl)piperazin-1-yl)-N-phenylacetamide derivatives 7a–n: (a) POCl3, DMF, 0˚C, then 80˚C, 12 h; (b) KI (10 mol%), K2CO3(1 eq), 80˚C, 2 h; (c) HCl, MeOH, RT, 3 h; (d) K2CO3(1 eq), DMF, 80˚C, 2 h; (e) thiosemicarbazide (1.5 eq), PTSA (20 mol%), DMF, 80˚C, 3 h.
Structure-activity relationships to target α-glucosidase
The SAR for this series of compounds based on α-glucosidase inhibitory activity reveals important insights into the effect of various substituents on the aryl moieties. The inhibitory potency, as reflected by the IC50 values, shows a wide range and specific trends can be identified (Table 1).
The unsubstituted phenyl ring exhibits moderate inhibitory activity (compound 7a, IC50 = 501.5 µM), serving as a baseline for comparison with substituted derivatives. Halogen substitution reveals important trends. Fluorine substitution of the phenyl ring, whether alone (compound 7b, 4-F) or in combination with chlorine (compound 7d, 3-Cl-4-F), leads to weak inhibitory activity. Fluorine is a small, highly electronegative atom that can influence electron distribution, but its size and low polarizability may reduce strong interactions with the enzyme. On the other hand, chlorine substitution at the 4-position (compound 7c) significantly enhances inhibitory activity (IC50 = 249.2 µM). Chlorine, being bulkier than fluorine and more polarizable, likely enhances hydrophobic and van der Waals interactions within the enzyme’s active site. This suggests that chlorine offers a more favorable balance of steric and electronic effects, improving binding compared to fluorine. The trifluoromethyl substitution on the phenyl ring (compound 7e) also shows good inhibition (IC50 = 389.0 µM). The electron-withdrawing nature of 4-CF3 contributes to enzyme interaction but may introduce steric hindrance due to its larger size compared to chlorine, which could explain its reduced potency relative to the 4-Cl derivative.
Nitro substitution demonstrates a position-dependent effect on enzyme inhibition. The nitro group at the 2-position of the phenyl ring (compound 7f) shows poor inhibitory activity (IC50 > 750 µM), likely due to steric clash within the enzyme active site. However, shifting the nitro group to the 3-position (compound 7g) significantly improves activity (IC50 = 309.4 µM).
Methyl substitution at the ortho position generally leads to diminished activity in compound 7h. Methyl group is electron-donating, and they do not contribute to hydrogen bonding. The absence of significant electronic effects or steric interactions likely explains the reduced activity in this case. Methoxy substitution shows position effects. Substitution with a methoxy group at the 4-position of the phenyl ring (compound 7i) results in poor inhibitory activity (IC50 > 750 µM). The methoxy group, being electron-donating, may increase electron density on the phenyl ring, but at the 4-position, it may not favorably interact with the enzyme’s active site. However, the di-substitution of methoxy groups at the 2- and 5-positions on the phenyl ring (compound 7j) significantly enhances inhibitory activity (IC50 = 50.0 µM). This compound showed 15-fold better activity vs. acarbose as the positive control. This dramatic improvement suggests that methoxy groups in these positions increase binding affinity.
Isoquinoline substitution (compound 7l) also shows moderate inhibitory potency (IC50 = 395.2 µM), reflecting improved activity compared to some phenyl derivatives. Isoquinoline introduces an additional nitrogen atom into the ring, which may engage in additional hydrogen bonding or electrostatic interactions with the enzyme.
The benzyl substitution (compound 7m) leads to weak inhibitory activity (IC50 > 750 µM). The 4-methoxy benzyl derivative (compound 7n) also exhibits poor activity, indicating that neither the larger size nor the electron-donating methoxy group favors binding. So it seems that in this case the length might increase the flexibility and rotation of molecules in the binding site of the enzyme and reduce the potency.
Enzyme kinetic studies
According to Fig. 2a, the Lineweaver-Burk plot showed that the Km gradually increased and Vmax remained unchanged with increasing inhibitor concentration indicating a competitive inhibition. The results show Compound 7j binds to the active site on the enzyme and competes with the substrate for binding to the active site. Furthermore, the plot of the Km versus different concentrations of the inhibitor gave an estimate of the inhibition constant, Ki of 32 µM (Fig. 2b).

Kinetics of α-glucosidase inhibition by 7j. (a) The Lineweaver– Burk plot in the absence and presence of different concentrations of 7j; (b) The secondary plot between Km and various concentrations of 7j.
Molecular docking study
Molecular docking is a powerful tool in drug discovery, used to simulate and analyze the interactions between ligand and its target receptor. It helps identify the key factors influencing the drug’s effectiveness against disease by visualizing how the ligand binds within the active site of the enzyme. In this study, molecular docking was performed on the homology-modeled α-glucosidase enzyme to gain insights into the binding interactions of the most potent analog. As expected, the most active compound, 7j, demonstrated several important interactions with the enzyme’s binding site, confirming its strong affinity (Fig. 3). Specifically, the quinoline ring of the analog formed two key pi-cation interactions with Arg312, which are crucial for stabilizing the ligand in the binding pocket. Additionally, a strong hydrogen bond interaction was observed between His279 and the sulfur atom of the thiosemicarbazide moiety, further enhancing the binding stability. Moreover, two additional hydrogen bond interactions were identified: one between the NH group of the amide linker moiety and Pro309, and the other between the carbonyl group (C = O) of the amide and Asn241. These interactions play a significant role in securing the ligand within the enzyme’s active site. The terminal 2,5-methoxyphenyl group of the molecule exhibited a pi-pi stacking interaction with Phe231, which further contributes to the stability of the enzyme-ligand complex. Each part of the molecule, from the quinoline core to the terminal substituted phenyl group, participates in distinct interactions, confirming the high affinity of the compound for the enzyme.

A docking model of α-glucosidase-7j complex (two- and three-dimensional interactions).
Molecular dynamic simulation
Molecular dynamic (MD) simulations were conducted to study the behavior of the 7j-enzyme complex over 300 ns in a simulated biological environment. This approach allows for the evaluation of the stability and conformational changes in the complex under dynamic conditions. Root Mean Square Deviation (RMSD) analysis was used to monitor structural deviations and stability throughout the simulation. RMSD is an essential metric that reflects how much the structure of the enzyme or complex changes relative to its initial configuration during the simulation. Generally, an RMSD (root mean square deviation) below 3 Å is considered acceptable, indicating that the protein remains close to its average thermal structure. For the α-glucosidase-7j complex, the RMSD remained below this threshold, suggesting that the system was stable and well-equilibrated during the simulation. Moreover, after 180 ns of simulation, the 7j-enzyme complex exhibited an even lower RMSD value of less than 2 Å, further confirming its stability. This low deviation suggests that the 7j forms a stable interaction with the enzyme, maintaining the overall conformation of the complex with minimal structural changes. In contrast, when the enzyme was analyzed without the ligand, a larger conformational shift was observed, with the RMSD reaching approximately 4.5 Å. (Fig. 4)

RMSD plot of α-glucosidase backbone (blue) and compound 7j (red), throughout the 300 ns of the simulation time.
The root mean square fluctuation (RMSF) of the backbone atoms in the 7j-enzyme complex was calculated to evaluate the flexibility of the complex. RMSF analysis provides insights into the mobility of specific regions of the enzyme, focusing on the fluctuations of α-glucosidase residues in the presence of the ligand. This analysis aims to determine how ligand binding affects the stability of various enzyme regions, particularly the active site and the A ___domain.
The results revealed that compound 7j significantly interacted with the B ___domain, active site, and A ___domain, leading to reduced fluctuations in these key regions (Fig. 5a). This reduction in flexibility suggests that the intermolecular interactions between the enzyme and ligand are robust, effectively restricting movement and stabilizing the enzyme’s structure. Furthermore, the decreased flexibility in the active site indicates that the enzyme-ligand complex is highly stable, reinforcing the potential of 7j as a potent α-glucosidase inhibitor.

(a) RMSF graph of the Cα atoms of α-glucosidase in complex with 7j and enzyme, (b) Interactions that happened for over 25% of the simulation.
The stability of the enzyme-ligand complex can be attributed to several key interactions between compound 7j and α-glucosidase (Fig. 5b). The quinoline moiety of 7j plays a central role by forming hydrogen bond with Arg312. The piperazine acetamide group of 7j is also involved in important interactions, forming hydrogen bonds with His239, which helps to maintain the ligand within the enzyme’s binding pocket. Furthermore, the amine group attached to 2,5-dimethoxyphenyl group of 7j forms an additional hydrogen bond with Ser308, adding to the overall stability of the enzyme-ligand complex. Lastly, the NH group of the thiosemicarbazide moiety forms two hydrogen bonds with Thr215 and Glu276 further reinforcing the interactions between the ligand and the enzyme. These multiple and diverse interactions—hydrogen bonding, pi-pi stacking, and hydrophobic contacts—create a highly stable complex that restricts the movement of key enzyme residues, as evidenced by the RMSF analysis. This highlights the potential of compound 7j as a strong α-glucosidase inhibitor, effectively stabilizing the enzyme and reducing its activity.
Hydrogen bonding is one of the most critical interactions for stabilizing protein-ligand complexes. The number of hydrogen bond interactions between compound 7j and the protein residues over the simulation time was also monitored. As shown in Fig. 6, in most frames, 7j successfully participated in hydrogen bond interactions, confirming its high potency.

Number of hydrogen bond interactions formed during MD simulation.
Additionally, the stability of the enzyme-ligand complex was further confirmed through the evaluation of the RMSF of the ligand (compound 7j) alone within the enzyme’s binding site, as shown in (Fig. 7). The RMSF values for all atoms of the ligand remained below 3 Å, indicating minimal fluctuation and confirming the stability of the ligand in the binding pocket. However, slight deviations were observed in atom 36, which exhibited slightly higher fluctuations compared to the rest of the ligand atoms. Despite these localized increases in RMSF, the overall low fluctuation of the majority of 7j confirms its stable binding, which demonstrated strong interactions between compound 7j and the critical residues of α-glucosidase.

RMSF of 7j within enzyme active site throughout the 300 ns of the simulation time.
Comparison between docking and MD simulations
The comparison between docking and MD simulations also highlights the dynamic and adaptive nature of protein-ligand interactions, emphasizing the importance of MD simulations for capturing realistic binding behaviors. As shown in Table 2, key residues such as Arg312 and His279 consistently participated in strong interactions during both docking and MD simulations, confirming their pivotal role in stabilizing the ligand within the active site. Residues like Thr215, His239, Glu276, and Ser308 formed hydrogen bond or water bridge interactions during MD simulations that were absent in docking results, highlighting the significance of accounting for dynamic water-mediated effects and conformational flexibility. In contrast, residues such as Phe231 and Pro309 exhibited interactions in docking but showed reduced persistence during MD simulations, indicating that these interactions might be weaker or context-dependent under dynamic conditions. Additionally, interactions involving residues like Asn241 and Glu276 demonstrated a combination of hydrophobic and water-mediated contributions, reinforcing their role in ligand stabilization. This analysis demonstrates the complementary strengths of docking and MD simulations in providing a comprehensive understanding of protein-ligand interactions, offering valuable insights for further optimization of the ligand’s design for enhanced inhibitory activity and stability.
Conclusion
In this study, we synthesized and evaluated a series of substituted piperazine conjugated to quinoline-thiosemicarbazide for their α-glucosidase inhibitory activity. The SAR analysis revealed that substitutions at specific positions on the aryl moieties significantly influenced inhibitory potency. Among the synthesized compounds, compound 7j (2,5-dimethoxyphenyl derivative) exhibited the most potent inhibitory activity, with an IC50 value of 50.0 µM, demonstrating a 15-fold improvement compared to the standard acarbose. Kinetic studies indicated that compound 7j acts as a competitive inhibitor, as confirmed by the Lineweaver-Burk plot, with a Ki value of 32 µM. Molecular docking and MD simulations provided further insight into the binding interactions between the compound and the α-glucosidase enzyme. Key interactions, including hydrogen bonding, pi-pi stacking, and hydrophobic interaction, contributed to the stabilization of the enzyme-7j complex. The low RMSD and RMSF values observed in the MD simulations confirmed the stability of the complex, supporting the potential of these compounds as strong inhibitors.
Overall, the findings suggest that thiosemicarbazide-linked quinoline-piperazine derivatives, particularly compound 7j, are promising candidates for the development of novel α-glucosidase inhibitors. These results provide valuable insights for further optimization and development of potent antidiabetic agents with improved efficacy and reduced side effects.
Experimental
Chemistry
Organic chemicals and solvents were sourced from Merck and used without additional purification. Analytical TLC was conducted with pre-coated aluminum sheets and silica gel to check reaction completion and compound purity. Melting points were measured using an Electrothermal 9100 apparatus. 1H NMR and 13C NMR spectra were recorded on Bruker 600 MHz instruments with TMS as the internal standard and CDCl3 as solvents.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-phenylacetamide
Yellow solid; Yield= (91%, 122.8 mg); mp: 135–137 °C. 1H NMR (600 MHz, CDCl3)δ 11.27 (s, 1 H), 9.80 (s, 1 H), 8.97 (s, 1 H), 8.44–8.06 (m, 3 H), 7.86–7.62 (m, 5 H), 7.48–7.24 (m, 3 H), 7.07 (s, 1 H), 3.29–3.12 (m, 5 H), 2.98–2.67 (m, 5 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 168.6, 159.7, 147.0, 139.2, 139.0, 135.8, 130.6, 129.1, 128.5, 127.6, 125.4, 125.2, 123.9, 121.5, 119.9, 62.2, 53.0, 50.9. HRMS (ESI)m/z: Calcd for C23H25N7OS [M + H] + 448.1920, Found 448.1927. FT-IRδ 3385, 3253, 1641, 1471, 773, 732 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(4-fluorophenyl)acetamide
Yellow solid; Yield= (84%, 117.2 mg); mp: 141–143 °C. 1H NMR (600 MHz, CDCl3)δ 11.72 (s, 1 H), 9.88 (s, 1 H), 8.97 (s, 1 H), 8.42–8.09 (m, 3 H), 7.89–7.58 (m, 5 H), 7.51–7.41 (m, 1 H), 7.24–7.06 (m, 2 H), 3.26 (s, 6 H), 2.91–2.72 (m, 4 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 168.6, 159.7, 159.3, 157.7, 147.0, 139.3, 135.4, 130.6, 128.5, 127.6, 125.4, 125.2, 121.9, 121.8, 121.5, 115.7, 115.6, 62.2, 53.0, 50.8. HRMS (ESI)m/z: Calcd for C23H24FN7OS [M + H] + 466.1825, Found 466.1819. FT-IRδ 3419, 3275, 1631, 1475, 1155, 771. 720 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(4-chlorophenyl)acetamide
Yellow solid; Yield= (81%, 116.9 mg); mp: 142–144 °C. 1H NMR (600 MHz, CDCl3)δ 11.71 (s, 1 H), 10.04 (s, 1 H), 8.99 (s, 1 H), 8.56–8.02 (m, 3 H), 7.87–7.25 (m, 8 H), 3.15–2.60 (m, 9 H), 1.23 (s, 1 H). 13C NMR (151 MHz, CDCl3)δ 178.5, 158.8, 146.8, 145.8, 139.0, 138.3, 137.4, 135.9, 130.8, 129.2, 128.5, 127.6, 125.9, 125.5, 121.6, 121.3, 52.6, 29.4, 21.2. HRMS (ESI)m/z: Calcd for C23H24ClN7OS [M + H] + 482.1530, Found 482.1521. FT-IRδ 3330, 3279, 1634, 1449, 830, 734, 720 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(3-chloro-4-fluorophenyl)acetamide
Yellow solid; Yield= (90%, 134.8 mg); mp: 140–142 °C. 1H NMR (600 MHz, CDCl3)δ 11.72 (s, 1 H), 10.06 (s, 1 H), 8.96 (s, 1 H), 8.49–7.19 (m, 10 H), 3.17 (s, 6 H), 2.82 (s, 4 H). 13C NMR (151 MHz, CDCl3)δ 178.5, 168.9, 159.7, 154.5, 152.8, 147.0, 139.3, 136.3, 135.8, 130.6, 128.5, 127.6, 125.4, 125.2, 121.5, 120.4, 120.3, 119.5, 119.4, 117.3, 117.1, 62.0, 53.0, 50.7. HRMS (ESI)m/z: Calcd for C23H23ClFN7OS [M + H] + 500.1436, Found 500.1426. FT-IRδ 3420, 3270, 1643, 1468, 1159, 835, 771, 725 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(4-(trifluoromethyl)phenyl)acetamide
Yellow solid; Yield= (90%, 139.1 mg); mp: 144–146 °C. 1H NMR (600 MHz, CDCl3)δ 11.71 (s, 1 H), 10.24 (s, 1 H), 8.99 (s, 1 H), 8.35 (s, 1 H), 8.26 (s, 1 H), 8.15 (s, 1 H), 7.98–7.67 (m, 7 H), 7.53–7.37 (m, 1 H), 3.26 (s, 5 H), 2.89 (m, 3 H), 2.73 (s, 2 H), 1.31–1.17 (m, 2 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 162.7, 146.9, 142.5, 139.2, 135.8, 130.7, 128.5, 127.6, 126.5, 125.7, 125.4, 125.3, 123.9, 121.5, 119.8, 52.9, 36.2, 31.2. HRMS (ESI)m/z: Calcd for C24H24F3N7OS [M + H] + 516.1793, Found 516.1799. FT-IRδ 3390, 3267, 1630, 1485, 1156, 793, 741 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(2-nitrophenyl)acetamide
Yellow solid; Yield= (85%, 125.5 mg); mp: 130–132 °C. 1H NMR (600 MHz, CDCl3)δ 11.73 (s, 1 H), 11.61 (s, 1 H), 8.99 (s, 1 H), 8.65 (s, 1 H), 8.46–8.06 (m, 4 H), 7.96–7.56 (m, 4 H), 7.53–7.19 (m, 2 H), 3.17 (s, 5 H), 3.07–2.60 (m, 5 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 170.2, 162.7, 159.7, 147.0, 139.3, 137.6, 136.1, 135.8, 134.0, 130.6, 128.5, 127.6, 126.1, 125.4, 124.1, 122.2, 121.5, 62.1, 53.2, 51.0. HRMS (ESI)m/z: Calcd for C23H24N8O3S [M + H] + 493.1770, Found 493.1778. FT-IRδ 3392, 3296, 1619, 1530, 1479, 1319, 780, 731 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(3-nitrophenyl)acetamide
Yellow solid; Yield= (80%, 118.1 mg); mp: 130–132 °C. 1H NMR (600 MHz, CDCl3)δ 11.69 (s, 1 H), 10.43 (s, 1 H), 9.01 (s, 1 H), 8.71 (s, 1 H), 8.44–8.08 (m, 3 H), 8.07–7.89 (m, 2 H), 7.88–7.73 (m, 2 H), 7.66 (s, 2 H), 7.52–7.40 (m, 1 H), 3.26–2.66 (m, 9 H), 2.27 (s, 1 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 162.7, 158.3, 148.4, 146.9, 146.1, 139.9, 139.1, 138.0, 135.9, 130.8, 128.5, 127.6, 126.0, 125.5, 121.4, 118.7, 114.2,52.8, 36.2, 31.2. HRMS (ESI)m/z: Calcd for C23H24N8O3S [M + H] + 493.1770, Found 493.1777. FT-IRδ 3383, 3279, 1655, 1529, 1476, 1310, 761, 738 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(o-tolyl)acetamide
Yellow solid; Yield= (89%, 123.1 mg); mp: 135–137 °C. 1H NMR (600 MHz, CDCl3)δ 11.74 (s, 1 H), 9.46 (s, 1 H), 8.98 (s, 1 H), 8.56–8.05 (m, 3 H), 7.99–6.89 (m, 8 H), 3.27 (s, 6 H), 2.86 (s, 4 H), 2.26 (s, 3 H). 13C NMR (151 MHz, CDCl3)δ 178.5, 168.1, 159.6, 147.0, 139.4, 136.5, 135.8, 130.7, 130.6, 129.5, 128.5, 127.6, 126.6, 125.4, 125.2, 124.9, 122.7, 121.5, 61.9, 53.2, 51.0, 17.9. HRMS (ESI)m/z: Calcd for C24H27N7OS [M + H] + 462.2076, Found 462.2083. FT-IRδ 3432, 3257, 1660, 1473, 783, 713 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(4-methoxyphenyl)acetamide
Brown solid; Yield= (93%, 133.1 mg); mp: 150–152 °C. 1H NMR (600 MHz, CDCl3)δ 11.67 (s, 1 H), 9.94 (s, 1 H), 9.02 (s, 1 H), 8.46–8.07 (m, 3 H), 7.99–7.41 (m, 6 H), 9.92 (d, J = 8.4 Hz, 2 H), 3.73 (s, 3 H), 3.43 (s, 8 H), 2.89 (s, 1 H), 2.73 (s, 1 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 162.7, 156.0, 146.8, 139.0, 135.9, 131.8, 130.7, 128.5, 127.6, 125.6, 125.5, 125.4, 121.5, 121.4, 114.3, 55.6, 52.7, 36.2, 31.2. HRMS (ESI)m/z: Calcd for C24H27N7O2S [M + H] + 478.2025, Found 478.2033. FT-IRδ 3420, 3246, 1649, 14,772, 1231, 1028, 785, 759 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(2,5-dimethoxyphenyl)acetamide
Brown solid; Yield= (84%, 127.8 mg); mp: 139–141 °C. 1H NMR (600 MHz, CDCl3)δ 11.72 (s, 1 H), 9.80 (s, 1 H), 8.99 (s, 1 H), 8.57–8.06 (m, 3 H), 8.05–7.36 (m, 5 H), 6.98 (s, 1 H), 6.62 (s, 1 H), 3.98–3.62 (m, 6 H), 3.28–2.65 (m, 10 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 168.3, 159.6, 153.6, 146.9, 142.7, 140.7, 139.2, 135.8, 130.6, 128.5, 127.6, 125.4, 125.3, 121.5, 112.1, 107.8, 106.1, 61.9, 57.0, 55.8, 53.1, 51.3. HRMS (ESI)m/z: Calcd for C25H29N7O3S [M + H] + 508.2131, Found 508.2139. FT-IRδ 3410, 3279, 1650, 1489, 1230, 1035, 789. 721 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(3,4-dimethoxyphenyl)acetamide
Brown solid; Yield= (88%, 133.9 mg); mp: 149–151 °C. 1H NMR (600 MHz, CDCl3)δ 11.70 (s, 1 H), 9.70 (s, 1 H), 9.00 (s, 1 H), 8.52-8.00 (m, 3 H), 7.93–7.57 (m, 3 H), 7.50–6.84 (m, 4 H), 4.00-3.64 (m, 6 H), 2.88 (s, 8 H), 1.36–0.75 (m, 2 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 149.0, 146.9, 145.5, 139.1, 138.0, 135.8, 132.5, 130.7, 128.6, 128.5, 127.6, 125.9, 125.4, 121.4, 112.4, 111.9, 106.1, 56.1, 55.8, 52.9, 31.4, 22.5. HRMS (ESI)m/z: Calcd for C25H29N7O3S [M + H] + 508.2131, Found 508.2122. FT-IRδ 3380, 3252, 1636, 1479, 1220, 1033, 791, 766 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(isoquinolin-5-yl)acetamide
Yellow solid; Yield= (87%, 130.0 mg); mp: 141–143 °C. 1H NMR (600 MHz, CDCl3)δ 11.73 (s, 1 H), 10.20 (s, 1 H), 9.35 (s, 1 H), 8.99 (s, 1 H), 8.58 (s, 1 H), 8.44–7.30 (m, 11 H), 3.42 (s, 6 H), 3.20–2.76 (m, 4 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 159.6, 153.1, 147.0, 143.3, 139.2, 135.8, 132.8, 130.7, 130.1, 129.1, 128.5, 127.7, 127.6, 125.9, 125.4, 125.3, 125.1, 125.0, 121.5, 115.6, 53.1, 50.7, 21.2. HRMS (ESI)m/z: Calcd for C26H26N8OS [M + H] + 499.2029, Found 499.2022. FT-IRδ 3430, 3210, 1666, 1471, 785, 711 cm− 1.
N-benzyl-2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)acetamide
Yellow solid; Yield= (91%, 126.0 mg); mp: 138–140 °C. 1H NMR (600 MHz, CDCl3)δ 11.72 (s, 1 H), 8.97 (s, 1 H), 8.70–7.98 (m, 4 H), 7.97–7.04 (m, 9 H), 4.34 (s, 2 H), 3.15 (s, 6 H), 2.76 (s, 4 H). 13C NMR (151 MHz, CDCl3)δ178.5, 169.6, 159.7, 147.0, 140.1, 139.4, 135.7, 130.6, 128.7, 128.5, 127.6, 127.1, 126.0, 125.4, 125.1, 121.5, 61.7, 53.2, 50.8, 42.4. HRMS (ESI)m/z: Calcd for C24H27N7OS [M + H] + 462.2076, Found 462.2071. FT-IRδ 3393, 3270, 1652, 1491, 789 cm− 1.
2-(4-(3-((2-carbamothioylhydrazono)methyl)quinolin-2-yl)piperazin-1-yl)-N-(4-methoxybenzyl)acetamide
Brown solid; Yield= (80%, 117.9 mg); mp: 143–145 °C. 1H NMR (600 MHz, CDCl3)δ 11.68 (s, 1 H), 8.99 (s, 1 H), 8.57–8.02 (m, 4 H), 7.93–7.60 (m, 3 H), 7.53–7.38 (m, 1 H), 7.32–7.10 (m, 2 H), 7.00-6.77 (m, 2 H), 4.27 (s, 2 H), 3.73 (s, 3 H), 3.28–2.61 (m, 10 H). 13C NMR (151 MHz, CDCl3)δ 178.4, 169.5, 159.7, 158.6, 147.0, 139.2, 135.2, 132.1, 130.6, 129.0, 128.5, 127.6, 125.4, 125.2, 121.5, 114.1, 61.7, 55.5, 53.2, 50.8, 41.8. HRMS (ESI)m/z: Calcd for C25H29N7O2S [M + H] + 492.2182, Found 492.2173. FT-IRδ 3386, 3277, 1679, 1480, 1229, 1051, 790, 735 cm− 1.
In vitro α-glucosidase inhibition assays
In vitro inhibition assay of the new compounds, 7a–n was performed according to previously reported work11,23.
Enzyme kinetic studies
The mode of inhibition of the most active compound (7j), identified with the lowest IC50, was investigated against α-glucosidase activity with different concentrations of p-nitrophenyl α-D-glucopyranoside (1–4 mM) as substrate in the absence and presence of 7j at different concentrations (0, 12.5, 25, and 50, 100 µM). A Lineweaver–Burk plot was generated to identify the type of inhibition and the Michaelis–Menten constant (Km) value was determined from the plot between the reciprocal of the substrate concentration (1/[S]) and the reciprocal of enzyme rate (1/V) over various inhibitor concentrations. The experimental inhibitor constant (Ki) value was constructed by secondary plots of the inhibitor concentration [I] versus Km24,25.
Docking study
Molecular modeling of the selected compound was conducted on modeled α-glucosidase based on our previously reported works26. The induced molecular docking was conducted using the Schrödinger Suites Maestro molecular modeling platform.
Molecular dynamics simulations
The starting model was obtained by imposing the induced fit docking to α-glucosidase. MD simulations were conducted using the Desmond v5.3 of Schrodinger’s suit maestro according to previously reported procedures23,27,28.
Data availability
All data generated or analyzed during this study are included in the published article and its supplementary information file.
References
Banday, M. Z., Sameer, A. S. & Nissar, S. Pathophysiology of diabetes: an overview. Avicenna J. Med. 10 (4), 174–188 (2020).
Antar, S. A. et al. Diabetes mellitus: Classification, mediators, and complications; a gate to identify potential targets for the development of new effective treatments. Biomed. Pharmacother. 168, 115734 (2023).
Eizirik, D. L., Pasquali, L. & Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat. Rev. Endocrinol. 16 (7), 349–362 (2020).
Galicia-Garcia, U. et al. Pathophysiology of type 2 diabetes mellitus. Int. J. Mol. Sci. 21 (17). (2020).
Sohrabi, M. et al. A review on α-glucosidase inhibitory activity of first row transition metal complexes: a futuristic strategy for treatment of type 2 diabetes. RSC Adv. 12 (19), 12011–12052 (2022).
Hossain, U. et al. An overview on the role of bioactive α-glucosidase inhibitors in ameliorating diabetic complications. Food Chem. Toxicol. 145, 111738 (2020).
van de Laar, F. A. et al. α-Glucosidase inhibitors for patients with type 2 diabetes: results from a cochrane systematic review and meta-analysis. Diabetes Care 28 (1), 154–163 (2005).
Zolotareva, D. et al. Morpholine, piperazine, and piperidine derivatives as antidiabetic agents. Molecules 29 (13), 3043 (2024).
Safapoor, S. et al. Synthesis, ADMT prediction, and in vitro and in silico α-glucosidase inhibition evaluations of new quinoline–quinazolinone–thioacetamides. RSC Adv. 13 (28), 19243–19256 (2023).
Abbasi, I. et al. Isatin-hydrazide conjugates as potent α-amylase and α-glucosidase inhibitors: synthesis, structure and invitro evaluations. Bioorg. Chem. 116, 105385 (2021).
Moghadam Farid, S. et al. Synthesis and structure–activity relationship studies of benzimidazole-thioquinoline derivatives as α-glucosidase inhibitors. Sci. Rep. 13 (1), 4392 (2023).
Noori, M. et al. Design, synthesis, and in silico studies of quinoline-based-benzo[d]imidazole bearing different acetamide derivatives as potent α-glucosidase inhibitors. Sci. Rep. 12 (1), 14019 (2022).
Ghomi, M. K. et al. Synthesis, in vitro potency of inhibition, enzyme kinetics and in silico studies of quinoline-based α-glucosidase inhibitors. Sci. Rep. 14 (1), 501 (2024).
Avula, S. K. et al. Synthesis of novel substituted quinoline derivatives as diabetics II inhibitors and along with their in-silico studies. J. Mol. Struct. 1274, 134560 (2023).
Elebiju, O. F. et al. Recent advances in functionalized quinoline scaffolds and hybrids-exceptional pharmacophore in therapeutic medicine. Front. Chem. 10, 1074331 (2022).
Lee, H. W., Yang, J. Y. & Lee, H. S. Quinoline-2-carboxylic acid isolated from ephedra pachyclada and its structural derivatives show inhibitory effects against α-glucosidase and α-amylase. J. Korean Soc. Appl. Biol. Chem. 57 (4), 441–444 (2014).
Taha, M. et al. Synthesis of quinoline derivatives as diabetic II inhibitors and molecular docking studies. Bioorg. Med. Chem. 27 (18), 4081–4088 (2019).
Rahim, F. et al. Isatin based thiosemicarbazide derivatives as potential inhibitor of α-glucosidase, synthesis and their molecular docking study. J. Mol. Struct. 1222, 128922 (2020).
Basri, R. et al. Synthesis, biological evaluation and molecular modelling of 3-Formyl-6-isopropylchromone derived thiosemicarbazones as α-glucosidase inhibitors. Bioorg. Chem. 139, 106739 (2023).
Saeedian Moghadam, E. et al. Synthesis, bioactivity, and molecular docking of benzimidazole-2-carbamate derivatives as potent α-glucosidase inhibitors. J. Mol. Struct. 1278, 134931 (2023).
Özil, M. et al. Synthesis of benzimidazoles containing piperazine ring as potential therapeutic agents against diabetes mellitus and antioxidant activities. J. Mol. Struct. 1304, 137714 (2024).
Ghasemi, M. et al. Quinoline-piperazine derivatives as potential α-glucosidase inhibitors: synthesis, biological evaluation, and in silico studies. J. Mol. Struct. 1323, 140561 (2025).
Iraji, A. et al. Cyanoacetohydrazide linked to 1,2,3-triazole derivatives: a new class of α-glucosidase inhibitors. Sci. Rep. 12 (1), 8647 (2022).
Hamedifar, H. et al. Aryl-quinoline-4-carbonyl hydrazone bearing different 2-methoxyphenoxyacetamides as potent α-glucosidase inhibitors; molecular dynamics, kinetic and structure–activity relationship studies. Sci. Rep. 14 (1), 388 (2024).
Nasli Esfahani, A. et al. Design and synthesis of phenoxymethybenzoimidazole incorporating different aryl thiazole-triazole acetamide derivatives as α-glycosidase inhibitors. Mol. Divers. 26 (4), 1995–2009 (2022).
Pedrood, K. et al. Design, synthesis, and molecular docking studies of diphenylquinoxaline-6-carbohydrazide hybrids as potent α-glucosidase inhibitors. BMC Chem. 16 (1), 57 (2022).
Azmi, A. et al. Alpha-glucosidase inhibitory and hypoglycemic effects of imidazole-bearing thioquinoline derivatives with different substituents: in silico, in vitro, and in vivo evaluations. Bioorg. Chem. 144, 107106 (2024).
Asadi, M. et al. Synthesis, α-glucosidase inhibitory activity, and molecular dynamic simulation of 6-chloro-2-methoxyacridine linked to triazole derivatives. Sci. Rep. 14 (1), 17338 (2024).
Acknowledgements
The authors wish to thank the support of the Vice-Chancellor for Research of Shiraz University of Medical Sciences (Grant Number: IR.SUMS.REC.1403.292).
Author information
Authors and Affiliations
Contributions
Author contribution M. G, M. M, M. D, and M. E synthesized compounds, S. M, and M. A. F performed biological assessments, A.I supervise the biological procedures, A. Al-H supervises the synthetic procedures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ghasemi, M., Mahdavi, M., Dehghan, M. et al. Substituted piperazine conjugated to quinoline-thiosemicarbazide as potent α-glucosidase inhibitors to target hyperglycemia. Sci Rep 15, 1871 (2025). https://doi.org/10.1038/s41598-024-83917-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-83917-z
Keywords
This article is cited by
-
Hybrid-based design and biological evaluation of quinoline-benzoylhydrazine based derivatives as α-glucosidase inhibitors
Medicinal Chemistry Research (2025)
