
Abstract
Crossbreeding in India has been widely adopted to address low sustainability and poor productivity in non-descript cattle. This study analyzed Vrindavani (VRI) crossbred cattle and their parental populations (Holstein Friesian (HOL), Jersey (JER), Brown Swiss (BSW), Hariana (HAR) using SNP data to characterize locus-specific ancestry in VRI’s genome along with admixture proportions and population stratification. Admixture analysis showed VRI have 67.3% HOL, 20.1% HAR, 8.5% JER, and 4% BSW ancestry. Locus-specific ancestry estimation identified regions with adaptive admixtures, which can be defined as admixed genomic regions favored by evolutionary forces and increased their frequencies, revealed 79.7% Bos taurus and 20.3% Bos indicus ancestry. Notably, regions on chromosome (chr) 2, 3, 4, 5, 7, 10, 12, 13, 14, 16, 17, 19, 20, 21, 22, 23, and 24 were associated with disease resistance contributed by indicine ancestry and chr 1, 6, 9, 11, 15, 18, 27, and 28 related to production which were contributed by taurine ancestry. The study concluded that increased taurine ancestry contributes to higher milk yield in VRI crosses, while indicine ancestry confers disease resistance and adaptability to tropical climates. This comprehensive genomic analysis suggests that while taurine inheritance enhances milk yield, a balance with indicine traits is essential for resilience. Understanding locus-specific ancestry patterns can aid in refining breeding strategies by selectively promoting beneficial alleles. Future advancements in genomic tools may enable controlled inheritance of desirable traits, maximizing heterosis in structured breeding programs for sustainable cattle production.
Similar content being viewed by others
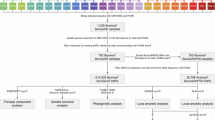


Introduction
Cattle ranked as the important domesticated animal in terms of economic impact, playing a key role in the development of human societies and civilizations. Genomic studies of domestic cattle have shown that cattle across the globe can be categorized into five main groups based on their continent of origin i.e., European taurine, Eurasian taurine, East Asian taurine, Chinese indicine, and Indian indicine1. Among them, Indian indicine has made a notable contribution to the global cattle genetic pool, with a total cattle population of 193.46 million. Of these, 51.36 million (26.5%) are well characterized and recognized indigenous breeds, while 142.11 million (73.5%) are non-descript or crossbred2. India also holds the title of the largest milk-producing nation, with a total milk production of 198.44 million metric tons in 2019-20, and cattle account for approximately 51% of this output3. Over the past years, interest on crossbreeding has increased in many countries due to its well documented advantages, largely attributed to heterosis or hybrid vigor and complementarity of the traits4,5. As a result, structured crossbreeding programs have been implemented in various nations, with Denmark, United States, and New Zealand leading the way6. In India, crossbreeding programs in cattle began in the 1970s, promoted by Indian Council of Agricultural Research (ICAR) and National Dairy Development Board (NDDB)7. Vrindavani (VRI) cattle is one such crossbred developed at the ICAR-Indian Veterinary Research Institute, by combining four breeds viz., Holstein Friesian (HOL), Hariana (HAR), Jersey (JER), and Brown Swiss (BSW)8,9.
Recent technological developments have enabled the cost-effective use of high-density DNA micro-arrays, allowing for the examination of thousands of genome-wide markers in all major livestock species10,11,13. This information has been employed in numerous studies to successfully trace fine-scale gene flow, identify selection signatures, and associate specific allelic variants with quantitative traits in various species14. Gene flow events, whether occurs from natural causes or human activities causes admixing of genomes. If such admixed genomic regions contribute adaptability to dynamic environment and genetic fitness, their frequency gets increased by natural or artificial selection. Such regions are known as adaptive introgression or adaptive admixtures15,16. High-resolution genome data has facilitated conceptualization of local ancestry inference methods that can determine the ancestry of specific chromosomal segments and all the chromosomal segments in the genome and pinpoints ancestry blocks17. When selection acts within an admixed population, the frequency of selected alleles tends to increase over several generations, leading to variations in locus-specific ancestry18. Consequently, adaptive introgression can be identified by detecting regions in the genome where ancestry is fixed or nearly fixed from a particular source population19. These deviations from expected patterns in the genomes of admixed individuals, whether excesses or deficiencies, serve as indicators of recent selective pressures. Because the effect on these regions accumulates over multiple generations, they can be interpreted as signs of selection following admixture19,20.
The examination of admixture signatures based on variations in local admixture levels has garnered significant interest in human18,19 and livestock genetic research21,22,23,24. In Indian cattle there are no locus-specific ancestral studies have been observed, hence to bridge this gap, the present investigation was taken to see the chromosome wide ancestry of the VRI crossbred cattle and impact of evolutionary forces.
Materials and methods
Vrindavani cattle (n = 96) genome wide markers were retrieved from25 Singh et al. (2020) available at https://figshare.com/ (https://doi.org/10.6084/m9.figshare.12808343.v2). Its parental populations i.e., HOL (n = 30), JER (n = 24), BSW (n = 21) and HAR (n = 10) genotyped using BovineSNP50 chip were obtained from WIDDE database.
Preparation of dataset and pruning for quality control
Genotype quality control (QC) was performed using PLINK v1.9 software26. All the variants with mapped coordinates in the UMD_3.1.1 bovine reference genome assembly ((https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000003055.6/)) were included for analysis. Only autosomal markers were considered and markers on sex chromosomes and mitochondrial DNA markers were removed in order to avoid any potential bias12,27. Markers with call rate (CR) exceeding 95% were kept for subsequent analysis. Individuals with missing genotype data with threshold (--mind) of 0.1 was also taken into consideration. Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) criteria were intentionally omitted, as they could potentially eliminate loci under selection and will influence the effects of population intermixing5,28,29,30,31.
Principal component and genome wide ancestry analysis
After QC analyses on the genomic dataset, Principal Component Analysis (PCA) was used for population structure study. PLINK v1.9 along with the ggplot package32 in R Studio was used for PCA and visualize its results33. To identify key components with the highest loadings for PC1 and PC2, we conducted Principal Component Analysis (PCA) using PLINK with variance-weighted SNP loadings (--pca var-wts). The SNP loadings from plink.eigenvec.var were extracted and ranked by absolute values to determine the top 20 SNPs contributing to PC1 and PC2. These SNPs were then mapped to their chromosomal positions using the .bim file. The identified SNPs were further analyzed for their association with known gene groups to assess their biological relevance using annotated UMD_3.1.1 genome reference assembly (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000003055.6/) with the help of in house developed tool called snp2feature (https://github.com/kkokay07/pq-genetics/tree/main/Annotation_of_features). Admixture analysis at genome level (global) was done to estimate ancestry proportions using ADMIXTURE software34. The analysis included 20,000 MCMC iterations following 10,000 burn-in steps. The optimal predefined number of ancestral groups (K) value, was determined by cross-validation errors. Admixture frequencies for all animals were calculated and plotted using Microsoft excel.
Inferring genome wide locus specific ancestry and adaptive admixtures
Local Ancestry in adMixed Populations (LAMP) is a software tool designed to estimate locus-specific ancestry in admixed individuals by analyzing the allele frequencies of reference populations35. Local ancestry for admixed animals (VRI in this study) was calculated using LAMP-ANC mode. In the LAMP configuration, a constant recombination rate of 1e-8 based on the assumption that 0.01 recombination’s occur per Mb (equivalent to 1 cM) was considered, given that no accurate genetic map is currently available for cattle. Furthermore, the excess and deficiency of local ancestry (‘Δ ancestry’) with respect to the reference panel breeds was estimated using the approach proposed by19,21. The ‘Δ ancestry’ captures significant shifts in ancestry variations across the genome and it is calculated by subtracting the genome-wide baseline ancestry from the average locus-specific ancestry for each of the two ancestral components19.
The Δ ancestry for ancestral population k at each SNP m is defined as:
\(\delta _{k}^{m}=\frac{1}{I}\mathop \sum \limits_{{i=1}}^{I} \left( {q_{k}^{{i,m}} - q_{k}^{{ - i}}} \right)\)
Where, \(q_{k}^{{i,m}}\)—locus-specific ancestry of animal i at SNP m; \(q_{k}^{{ - i}}\)—mean of the locus-specific ancestry for individual I.
Finally, the outlier approach is used to find adaptive admixtures from local ancestry information. The regions which exceeded the 3 ± SD (standard deviation) were considered as adaptive admixtures21. Finally, structural annotation was done using the snp2feature (https://github.com/kkokay07/pq-genetics/tree/main/Annotation_of_features) and genomic differences between these populations were established through the functional genomic studies by carrying out functional annotation using DAVID, g:Profiler36 and Animal QTL database37. Also, phenotypic annotation was done using OMIA (Online Mendelian Inheritance in Animals) (https://www.omia.org/home/).
Results
Prior to quality control (QC), the dataset comprised 181 individuals and 51,998 single nucleotide polymorphism (SNP) markers. Following QC procedures, approximately 9,553 SNPs were removed, resulting in the retention of 42,445 SNPs for downstream analyses. Principal component analysis revealed that the first two principal components effectively separated the data into distinct groups based on geographic origins. Taurine breeds viz., HOL, JER, and BSW, formed a distinct cluster, whereas the indicine breed, HAR, formed a separate cluster. The VRI cattle population was positioned between these two clusters but exhibited a greater affinity toward the taurine group, particularly HOL, indicating a higher proportion of taurine ancestry. The first and second principal components accounted for 20.27% and 11.833% of the total genetic variation, respectively (Fig. 1). The top 20 SNPs were identified across different chromosomes each for PC1 and PC2. Several SNPs were linked to genes with important functions (Table 1).

The principal component analysis (PCA) plot illustrates how various cattle breeds cluster based on their geographic locations. The first and second principal components explained 20.27% and 11.83% of the total variation, respectively. BSW-Brown Swiss; HAR-Hariana; HOL-Holstein Friesian; JER-Jersey; VRI-Vrindavani.
Genome wide ancestry estimation
The individual admixture proportions, estimated using the full set of SNPs for both parental and admixed populations, are presented in Fig. 2. The admixture analysis with K = 4 revealed the composition proportions of the parental populations—BSW, HOL, JER, and HAR—at 4%, 67.3%, 8.5%, and 20.1%, respectively (Supplementary file S1 and S2).

Individual-wise bar plot results for dataset for K value of 4, separating the populations/breeds from their lineages. BSW-Brown Swiss; HAR-Hariana; HOL-Holstein Friesian; JER-Jersey; VRI-Vrindavani.
Locus specific ancestry estimation
Locus-specific ancestry (local ancestry) estimation was conducted using the delta ancestry method by classifying all exotic breeds as part of the Bos taurus group (HOL, JER, and BSW) and HAR in the Bos indicus group. Regions exhibiting adaptive admixture were identified using an outlier approach, wherein loci exceeding 3 ± SD were considered significant. These noteworthy regions are highlighted in red (Figs. 3 and 4). Within the genomic landscape, adaptive admixtures contributed by Bos indicus were detected on Chr 2, 3, 4, 5, 7, 10, 12, 13, 14, 16, 17, 19, 20, 21, 22, 23, and 24. In contrast, those attributed to Bos taurus were localized to Chr 1, 6, 9, 11, 15, 18, 27, and 28.

Local ancestry analysis of chromosome 1–16 in Vrindavani cattle population. Portions above the green broken line in the middle of each plot are contributed by Bos indicus ancestry (Hariana cattle) and those below were contributed by Bos taurus ancestry (Holstein Friesian, Jersey and Brown Swiss). Those regions identified with red color are adaptive admixtures.

Local ancestry analysis of chromosome 17–29 in Vrindavani cattle population. Portions above the green broken line in the middle of each plot are contributed by Bos indicus ancestry (Hariana cattle) and those below were contributed by Bos taurus ancestry (Holstein Friesian, Jersey and Brown Swiss). Those regions identified with red color are adaptive admixtures.
Adaptive admixture with excess of indicine or taurine ancestry in Vrindavani cattle
Haplotypes of indicine and taurine ancestry may confer VRI a relative adaptive advantage under selection pressures. To identify haplotypes associated with selection in VRI cattle, an outlier approach was applied. The analysis identified 609 adaptive regions of indicine ancestry and 4,868 adaptive regions of taurine ancestry, encompassing approximately 534 and 4,321 genes, respectively (Supplementary file S6, S7, S9 and S10). The highest number of regions under selection from indicine ancestry (105 regions) was observed on chr 5, whereas the highest number from taurine ancestry (1,000 regions) was detected on chr 18, followed closely by chr 11 (845 regions) and chr 1 (826 regions) (Fig. 5). Gene ontology (GO) enrichment analysis revealed several biological processes and pathways associated with both indicine and taurine ancestry (Supplementary file S3 and S4).

Chromosome-wise distribution of genes in linkage disequilibrium with adaptive admixtures from Bos taurus and Bos indicus in Vrindavani cattle.
QTL enrichment analysis
A total of 69 and 107 quantitative trait loci (QTLs) were identified within the adaptive admixtures of VRI contributed by Bos indicus and Bos taurus, respectively, associated with exterior traits, production, reproduction, milk, and carcass traits (Supplementary file S5 and S8.
). The majority of these QTLs were of Bos taurus origin, constituting more than 50% in each category. For milk production Bos indicus QTLs accounted for approximately 51% (Fig. 6). Chromosome 1 and 14 harbored the most important QTLs, originating from Bos taurus and Bos indicus, respectively, with both predominantly associated with milk production. QTLs of taurine ancestry were distributed across chr 1, 6, 9, 11, 15, 18, 27, and 28, whereas those of indicine ancestry were located on chr 2, 3, 4, 5, 10, 12, 13, 14, 16, 19, 21 and 23. The chromosome-wise distribution of QTLs of taurine and indicine ancestry in Bos taurus and Bos indicus is presented in Fig. 7.

Proportion quantitative trait loci (QTL) related to different categories in linkage disequilibrium with adaptive admixtures in Vrindavani cattle contributed by Bos taurus and Bos indicus ancestry.

Chromosome-wise distribution of quantitative trait loci (QTL) in linkage disequilibrium with adaptive admixtures in Vrindavani cattle contributed by Bos taurus and Bos indicus ancestry.
Phenotype enrichment through online Mendelian inheritance in animal (OMIA)
The Online Mendelian Inheritance in Animals (OMIA) database (https://omia.org/home/) was queried to identify variants previously reported as causal or associated with cattle diseases, coat color, or other phenotypic traits. Genes play a crucial role in determining various observable traits across different organisms, and their functional implications in cattle are summarized in Supplementary file S11.
Functional enrichment analysis
The functional enrichment analysis of genes linked to adaptive admixtures in VRI cattle, derived from Bos indicus and Bos taurus ancestry, identified significant molecular functions (MF), biological processes (BP), and cellular components (CC) associated with environmental adaptation (Figs. 8 and 9). In the molecular functions category, the most significant term from Bos indicus ancestry were the structural constituent of the skin epidermis (GO:0030280, Padj = 5.99 × 10–21), transcription cis-regulatory region binding (GO:0000976) and DNA-binding transcription factor activity (GO:0003700). From Bos taurus ancestry, protein binding (GO:0005515, Padj = 7.71 × 10–7), sequence-specific double-stranded DNA binding (GO:1990837), transcription regulator activity (GO:0140110), catalytic activity (GO:0003824) and small molecule binding (GO:0036094) were significantly enriched.
In the biological processes category, Bos indicus-derived genes were significantly enriched in pathways related to keratinization (GO:0031424, Padj = 3.73 × 10–17), intermediate filament cytoskeleton organization (GO:0045104), system development (GO:0048731) and anterior/posterior pattern specification (GO:0009952). Bos taurus-derived genes showed significant enrichment in anatomical structure development (GO:0048856, Padj = 1.35 × 10–13), cell surface receptor signalling pathway (GO:0007166), plasma membrane-bounded cell projection organization (GO:0120026) and ovulation cycle process (GO:0022606).
In the cellular component category, Bos indicus-derived genes showed significant enrichment in keratin filament (GO:0045095, Padj = 7.13 × 10–19) and transcription regulator complex (GO:0005667). Bos taurus-derived genes were significantly enriched in extracellular matrix organization (GO:0030198, Padj = 3.54 × 10–3), response to lipid (GO:0071398), and cell adhesion (GO:0007155), cytoplasm (GO:0005737), nucleoplasm (GO:0005654), and cell junctions (GO:0030054, GO:0005911), very-low-density lipoprotein particles (GO:0034361) and extracellular vesicles (GO:0031012). These findings highlight the genetic contributions of both Bos indicus and Bos taurus ancestry in shaping adaptive traits in VRI cattle, with enriched pathways linked to thermotolerance, immune function, metabolic regulation, reproductive efficiency, and structural integrity.

Gene ontology enrichment of genes in linkage disequilibrium with the adaptive admixtures in Vrindavani cattle derived from Bos indicus ancestry.

Gene ontology enrichment of genes in linkage disequilibrium with the adaptive admixtures in Vrindavani cattle derived from Bos taurus ancestry.
Discussion
To address low sustainability and poor productivity, crossbreeding has been widely adopted in India. Despite its importance, the genomic attributes contributing to the advantages of crossbreeding have not been thoroughly explored across the entire genome. Previous studies on VRI cattle have mainly focused on various diversity parameters, neglecting the estimation of local ancestry. Vrindavani cattle is a composite breed8,9,25,38 designed to exploit breed complementarity and heterosis (hybrid vigor)16. Understanding the admixture proportions of individuals helps estimate heterozygosity, comprehend the breeding history of the population, and make informed management decisions for crossbreeding programs39. Historically, breed origins were estimated using microsatellite markers40 and more recently using SNPs8. DNA markers are accurate for estimating animal ancestry because they measure realized parental contributions at the genomic level41. This can correct pedigree errors and estimate kinships when pedigree data are incomplete or missing42,43.
Our study characterized admixed genome of VRI cattle using their parental genomes viz., HOL, BSW, JER and HAR. According to44, the sample size of our study is sufficient for admixture studies and aligns with previous research9,25,45. Only the markers present on autosomes are generally included in population genetic studies to remove the biases and ambiguities arising from sex chromosomes5. After quality control, 181 individuals and 42,445 SNPs left for subsequent analysis. The quality SNPs obtained were in agreement with other mid density genotyping viz.46,47,48, wherein 40,492, 54,404, and 54,609 SNPs were used, respectively, and were relatively higher compared to previous studies on VRI cattle8,25. This comparison suggests good quality genotypes used in the present study.
Population stratification
Using PCA, we observed that the first two principal components distinctly separated the data based on geographic origins. Taurine breeds (HOL, JER and BSW) formed one cluster, while the Indicine breed (HAR) formed another (Fig. 1). Interestingly, Vrindavani cattle was positioned between the taurine and indicine lineages, leaning more towards the taurine breeds as it can be attributed to their more than 50% taurine ancestry, consistent with previous findings8,25. Similarly, Frieswal cattle, a crossbred developed in India, exhibited a comparable pattern, positioned between the Taurine (HOL) and Indicine (Sahiwal) breeds, with an 61.5% taurine ancestry45. Also observed some individuals from the Red Sindhi breed clustering towards the JER breed49.
The PCA revealed significant SNPs associated with key genomic regions that potentially influence various biological functions in cattle. In PC1, the top 20 SNPs were identified with feature loading scores ranging from 2.73207 to 2.91285, emphasizing regions of functional relevance. The highest feature loading score (2.91285) was observed for SNP Hapmap27031-BTA-161,389 on chr 18, linked to PHKB, a gene crucial for glycogen metabolism and energy balance in muscle function50. Other associations include KCNN3 (chr 3: 15818013), which encodes a calcium-activated potassium channel involved in neural signaling and muscle contractility51, and PREX1 (chr 13: 77635899), which influences meat quality traits in Simmental cattle52. SRGAP2 (chr 16: 4061366) has been identified as a potential regulator of milk production in indicine cattle53. ADAMTS6 (chr 20: 14290727), a gene associated with extracellular matrix remodeling and skeletal muscle development, regulating myoblast proliferation and differentiation in cattle-yak54. Additionally, MIB1 (chr 24: 3476101) was associated with the fatty acid profile in Belgian Blue cattle55. TDGF1 (chr 22: 53470362) plays a critical role in embryonic development, the estrous cycle, and early pregnancy56, while LRRC2 (chr 22: 53470362) is involved in epigenetic modifications in skeletal muscle57.
In PC2, the top 20 SNPs were identified across several chromosomes, with feature loading scores ranging from 3.21094 to 3.87454. The highest feature loading score (3.87454) was observed for SNP ARS-BFGL-NGS-41,209 on chr 6, linked to the unannotated locus LOC107131184. Several SNPs were associated with genes playing essential roles in physiology, development, and adaptation. MAP7D1, TRAPPC3, and COL8A2 (chr 3: 11027602) are involved in cytoskeletal organization, vesicle trafficking, and extracellular matrix composition, which are vital for muscle structure and tissue integrity in cattle58,59,60. NUDCD3 (chr 4: 77635835) may play a role in cell division and developmental pathways61. PDGFRA (chr 6: 71421017) encodes a receptor involved in cell proliferation and differentiation, making it a strong candidate for growth traits in cattle62. H2AFY (chr 7: 4876442) encodes a histone variant crucial for chromatin remodeling and gene regulation, potentially impacting epigenetic modifications in cattle63. TBX18 (chr 9: 65564012) is involved in embryonic development, particularly in cardiovascular and urogenital system formation, with potential implications for reproductive traits. It has been associated with development in the dual-purpose Simmental breed but not in other specialized breeds64. TP53BP2 (chr 16: 27894801) plays a critical role in apoptosis and cell cycle control, suggesting its potential involvement in immune response and disease resistance65. Another significant gene, GRM7 (chr 22: 19168846), a metabotropic glutamate receptor, may influence nervous system function and behavioral traits in cattle62.
Genome-wide ancestry estimation
Admixture analysis using parental populations determined the ancestry proportions in VRI cattle at K = 4 (Fig. 2). The genetic contributions were identified as 67.3% from HOL, 8.5% from JER, 4% from BSW and 20.1% from HAR, indicating a strong taurine ancestry. These findings are consistent with earlier studies on VRI cattle8,25 and Frieswal cattle38, though with different proportions which could be due to quality control parameters that we incorporated in our study, mainly the Hardy-Weinberg equilibrium and linkage disequilibrium criteria were not applied as they may remove loci under selection and also remove influence of intermixing of populations with different gene frequencies5,28,29,30. Similarly, crossbred cattle in Maharashtra, India, have an average exotic ancestry of 70.3%66. Additionally, one more study on Indian cattle showed that while breeds such as Sahiwal, Tharparkar, Gir, Ongole, Kangayam, and Hariana formed distinct genetic clusters; Vechur exhibited significant admixture, likely due to crossbreeding67. Higher levels of taurine ancestry might not be suitable for tropical climate, as optimal production and economic traits are generally observed at exotic inheritance levels between 50% and 62.5%. Higher levels of exotic inheritance often result in decreased performance in both production and reproduction traits68. Research on Sahiwal and Holstein crossbreds69 revealed no improvement in production beyond 50% exotic inheritance. Conversely, lower exotic inheritance in crossbred cattle suggests better adaptation to tropical conditions and less emphasis on selecting for exotic traits. Given that crossbred cows contribute to 54% of India’s total milk production3, it is crucial to address any discrepancies in inheritance levels by refining breeding strategies.
Locus-specific ancestry
Adaptive admixture regions, resulting from hybridization between distinct parental populations, were investigated to understand their role in evolutionary adaptation. These genomic segments emerge in hybrid progeny and, when conferring fitness advantages, are subject to positive selection. Regions exhibiting significantly elevated frequencies toward fixation were identified using an outlier detection approach21. Identifying distinct ancestral origins of genomic segments has numerous applications, such as boosting milk production, mapping diseases, and uncovering historical genetic events16,35. This allows us to detect beneficial local genetic traits from specific breeds in mixed livestock populations. For instance, genomic sequence analysis has shown that local ancestry segments from Angus, Brahman, and Wannan breeds might enhance rapid growth, immune resistance, and indigenous adaptation in Yunling cattle70. Furthermore, research has found evidence of Mito-nuclear co-evolution in hybrid African cattle populations, with a notable increase in taurine ancestry at nuclear genes targeted by mitochondria71.
Local ancestry estimation on Vrindavani cattle was performed using the delta ancestry method, classifying all exotic breeds (HOL, JER and BSW) and indicine breed (HAR) parental breed as part of the Bos taurus group and Bos indicus group respectively. Previously Local ancestry analysis has been effective in identifying recent selection in admixed populations, such as Swiss Fleckvieh cattle72 and Zebu introgressed regions in Colombian creole taurine cattle73. Used a similar approach to detect significant differences in local admixture levels, identifying five chromosomes with notable deviations from average ancestries74. This included an excess of Baoulé ancestry, potentially linked to higher trypanosomiasis tolerance, particularly on chr 6, 8, and 19 in trypanosome-negative individuals. Additionally, higher Baoulé ancestry on Chr 8 (35–50 Mb) in trypanosome-positive cattle suggests beneficial Baoulé haplotypes unrelated to trypanosomiasis tolerance. Similarly, on Chr 2, 5, 7, 12 and 14 of VRI cattle we found the genes and QTLs related to disease resistance which is attributed to the HAR breed (known for tropical adaptation). For instance, region on chr 7 of Vrindavani cattle overlaps with a previously reported region for Mycobacterium paratuberculosis susceptibility in U.S. Holsteins75 and encompasses the SPOCK1 gene, which has been shown to be associated with cancer in humans76. Discovered that genes such as VAV1, PIK3R5, RAC1, VAV2, GAB2, and INPP5D on Chr 8 are under selection in Muturu and N’Dama cattle breeds in response to trypanosome infection77.
In VRI cattle, chr 6, 9, 11, 15, 27, 28 harbored genes and QTLs related to milk production as these regions are mostly of taurine ancestry, while QTLs and genes on chr 2, 10 and 14 are contributed by the HAR breed. Because of higher level of taurine ancestry, the milk production in VRI cattle (9.9 kg/day) is usually high when compared to the HAR cattle (2.4Kg/day)7,78, suggesting VRI have been benefited of positive heterosis. Indicine alleles introgression also be responsible for increase milk yield, as their genes are involved in environmental adaptation that allows expression of the milk production potential of crossbred79.
Adaptive admixtures contributed by Bos indicus
The annotation of adaptive admixtures has unveiled intriguing genetic insights of Bos indicus in VRI cattle. YTHDC2 gene on chr 10 associated with the iron content in milk25. Chromosome 14 harbored the gene DGAT1, a well-known enzyme is responsible for production of milk triglycerides in HOL cattle80,81,82. Gene HSF1 on chr 14 binds to heat shock elements in HSP genes and is activated by stress, controls the transcription of HSP genes83 and this regulatory role is crucial under conditions of elevated temperature as the VRI cattle are in tropics. Similarly, TONSL and VPS28 genes on are involved in cellular response to heat stress, where VPS28 gene was involved in heat shock resistance while TONSL maintains genomic stability during increased heat conditions82,84,85. Polymorphisms on gene PLCB1 on Chr 13 is a candidate gene for milk production in Indicine cattle86 which is inherited to VRI cattle by HAR cattle.
Adaptive admixtures contributed by Bos taurus
Within the adaptive admixtures region attributed to Bos taurus on the Chr 1 in VRI cattle, genes like UMPS, ITGB2, IFT80, DGKG, CLDN16 and ATR which are related to disease resistance and genetic adaptations, were found. Chromosome 11 harbored genes related to reproduction like TTF1, SNAPC4, RPIA, and MED22. Chr 6 harbored important genes like KIT, HMX and PRKG2 genes. KIT gene is responsible for white spotting (coat color) in VRI cattle87. Also, variations in KIT gene are responsible for the coat color patterns in Hereford cattle, the spotting patterns in Holstein, Simmental cattle88, and Fleckvieh cattle89, as well as the degree of black coat color in Holstein cattle90. It also facilitates the migration of melanocytes from the neural crest to the skin, which is essential for melanogenesis91. HMX gene on chr 6 also plays a crucial in external ear development92,93. PRKG2 gene is responsible for growth and carcass traits in cattle94, chicken95 and has been reported in humans that PRKG2 gene deletion is associated with growth restriction96.
Other important genes having taurine ancestry were ADAM2, ADAM32, CHST8, ITGB5, PLAT, TGFA and UPK1B. These genes have been previously reported in genome wide association studies (GWAS) that are associated with reproductive traits97. APOB and STK33 gene were associated with milk production25, whereas DDX1 gene was found to be associated with the involution of bovine mammary glands under environmental stress98 and it was also been linked to viral resistance and the amount of linoleic acid in Nellore cattle99. Previously reported genes in Vrindavani cattle in selection signature studies25 were also detected in present study i.e., DENND5A, NBAS, NRIP3, SCUBE2, TRIM66.
As the milk production in VRI cattle was higher when compared to its indicine parent (HAR), several genes associated with the milk protein percentage (CENPN, CMIP, NECAB2, OSGIN1, CLNK), milk mineral like Copper (PPA2, PPP2R2C), iron (RHPN2, DPY19L3, FAAP24, LRP3) and Phosphorus (NAT10) were enriched. Other genes such as ALB and PDE9A involved in variation in milk constituents such as milk fat, protein and milk production respectively, were also reported in Holstein cattle and Murrah buffaloe100,101,102. Apart from KIT gene, BDNF was also responsible for coat coloration and was reported previously through GWAS studies in Vrindavani cattle87. ESR1 and FSHR gene previously identified in Gir and Tharparkar cattle, plays a crucial role in sexual development and fertility in livestock [14, 104, 86104, . ATP2C1 and CAPN5 had a substantial relationship with cattle meat tenderness, marbling score, and proteolytic activity in cells in Gir86 and Chinese Wagyu cattle105. As VRI cattle is tropical cattle, HSPB6 gene plays a crucial role as it was strongly correlated with thermo-tolerance in cattle breeds like Sahiwal, Gir and South African Zebu cattle86,106,107.
Functional enrichment of adaptive admixtures in Vrindavani cattle
The molecular functions enriched in Bos indicus-derived genes primarily reflect adaptations to environmental stressors, particularly thermotolerance and epidermal integrity. The significant enrichment of structural constituents of the skin epidermis (GO:0030280) highlights the critical role of keratin-associated proteins in protecting against heat stress, UV radiation, and parasite load108. The presence of transcription cis-regulatory region binding (GO:0000976) and DNA-binding transcription factor activity (GO:0003700) suggests a strong regulatory adaptation, likely facilitating gene expression changes in response to environmental cues109.
In contrast, Bos taurus-derived genes were significantly enriched for protein binding (GO:0005515) and sequence-specific double-stranded DNA binding (GO:1990837), indicating the importance of protein-protein interactions and regulatory networks in metabolic adaptation110. The enrichment of catalytic activity (GO:0003824) and small molecule binding (GO:0036094) further suggests a crucial role in enzymatic processes and ligand-mediated biochemical pathways, which are essential for metabolic efficiency, nutrient utilization, and immune resilience111. These findings suggest that Bos taurus ancestry enhances metabolic regulation, providing VRI cattle with improved physiological adaptability under variable environmental conditions.
Conclusion
The analysis of Vrindavani cattle revealed a predominantly taurine ancestry, with significant genetic contributions from Bos taurus breeds such as Holstein Friesian, Jersey, and Brown Swiss. However, the substantial taurine inheritance may not be optimal for tropical climates, where a balance between exotic and indigenous genetic traits is crucial. Local ancestry analysis highlighted adaptive admixtures from Bos indicus breeds, contributing to traits like disease resistance and environmental adaptation. Specific genomic regions and genes associated with these traits were identified, offering insights into the genetic basis of Vrindavani cattle’s performance. The findings highlight the importance of maintaining genetic diversity to optimize production and adaptation traits. Crossbreeding strategies should aim for heterozygosity to harness the benefits of both taurine and indicine ancestries. Currently, in a crossbreeding program, because of random recombination and independent assortment of genetic material, we can’t control the inheritance of desirable alleles from the different ancestry. However, by analyzing local ancestry we can get to know the inheritance patterns of alleles. In future, certain technology can be developed in order to control inheritance of desirable combinations of alleles, hence it may maximize heterosis in crossbreeding programs and also investigating how external environmental factors, particularly India’s diverse geographical landscapes and climatic conditions, influence the selective pressures on genes in Vrindavani cattle.
Data availability
The datasets presented in this study were retrieved from public repository/repositories and link was https://figshare.com/ (https://doi.org/10.6084/m9.figshare.12808343.v2).
References
Chen, N. et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 9 (1), 2337 (2018).
Department of Animal Husbandry and Dairying. Government of India, Annual report 2022- 23 (2023). https://dahd.nic.in/.
20th Livestock Census. BAHS-basic animal husbandry statistics: Department of Animal Husbandry, Dairying & Fisheries, Ministry of Agriculture, Government of India. www.dahd.nic.in (2019).
Sorensen, M. K., Norberg, E., Pedersen, J. & Christensen, L. G. Invited review: crossbreeding in dairy cattle: a Danish perspective. J. Dairy Sci. 91 (11), 4116–4128 (2008).
Kanaka, K. K. et al. On the concepts and measures of diversity in the genomics era. Curr. Plant Biol. 33, 100278 (2023).
Kargo, M., Madsen, P. & Norberg, E. Is crossbreeding only beneficial in herds with low management level? J. Dairy Sci. 95 (2), 925–928 (2012).
Singh, R. R., Dutt, T., Kumar, A., Tomar, A. K. S. & Singh, M. On-farm characterization of Vrindavani cattle in India. Indian J. Anim. Sci. 81 (3), 267–271 (2011).
Ahmad, S. F. et al. Revelation of genomic breed composition in crossbred cattle of India with the help of Bovine50K BeadChip. Genomics 112 (2), 1531–1535 (2020).
Chhotaray, S. et al. Ancestry informative markers derived from discriminant analysis of principal components provide important insights into the composition of crossbred cattle. Genomics 112 (2), 1726–1733 (2020).
Kijas, J. W., Hadfield, T., Sanchez, N., Cockett, N. & M., & Genome-wide association reveals the locus responsible for four‐horned ruminant. Anim. Genet. 47 (2), 258–262 (2016).
Sukhija, N. et al. The flight of chicken genomics and allied omics-a mini review.Ecol. Genet. Genom. 2023, 100201 (2023).
Sukhija, N. et al. Evolutionary stamps for adaptation traced in Cervus nippon genome using reduced representation sequencing. Conserv. Genet. Resourc. 16(1), 135–146 (2024).
Goli, R. C. et al. Unraveling the genetic tapestry of Indian chicken: a comprehensive study of molecular variations and diversity. Ecol. Genet. Genom. 2024, 100220 (2024).
Sukhija, N. et al. Genome-wide selection signatures address trait specific candidate genes in cattle indigenous to arid regions of India. Anim. Biotechnol. 35 (1), 2290521 (2024).
Hedrick, P. W. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 22 (18), 4606–4618 (2013).
Goli, R. C. et al. Global and local ancestry and its importance: a review. Curr. Genom. 25 (4), 237–260 (2024).
Tang, H., Coram, M., Wang, P., Zhu, X. & Risch, N. Reconstructing genetic ancestry blocks in admixed individuals.Am. J. Hum. Genet. 79 (1), 1–12. https://doi.org/10.1086/504302 (2006).
Bhatia, G. et al. Genome-wide scan of 29,141 African Americans finds no evidence of directional selection since admixture. Am. J. Hum. Genet. 95 (4), 437–444 (2014).
Tang, H. et al. Recent genetic selection in the ancestral admixture of Puerto Ricans. Am. J. Hum. Genet. 81, 626–633 (2007).
Long, J. C. The genetic structure of admixed populations. Genetics 127 (2), 417–428 (1991).
Gautier, M. & Naves, M. Footprints of selection in the ancestral admixture of a new world Creole cattle breed. Mol. Ecol. 20 (15), 3128–3143 (2011).
Flori, L. et al. Adaptive admixture in the West African bovine hybrid zone: insight from the Borgou population. Mol. Ecol. 23 (13), 3241–3257 (2014).
Kim, E. S. & Rothschild, M. F. Genomic adaptation of admixed dairy cattle in East Africa. Front. Genet. 5, 443 (2014).
Bahbahani, H. et al. Signatures of positive selection in East African Shorthorn Zebu: A genome-wide single nucleotide polymorphism analysis. Sci. Rep. 5(1), 1–13 (2015).
Singh, A. et al. Signatures of selection in composite Vrindavani cattle of India. Front. Genet. 11, 589496 (2020).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. j. Hum. Genet. 81 (3), 559–575 (2007).
Goli, R. C. et al. Insights from homozygous signatures of cervus nippon revealed genetic architecture for components of fitness. Mammalian Genome 35(4), 657–672 (2024).
Falconer, D. S. Introduction To Quantitative Genetics (Pearson Education India, 1996).
Laird, N. M. & Lange, C. The Fundamentals of Modern Statistical Genetics 167 (Springer, 2011).
Abramovs, N., Brass, A. & Tassabehji, M. Hardy-Weinberg equilibrium in the large-scale genomic sequencing era. Front. Genet. 11, 210 (2020).
Mahar, K. et al. Runs of homozygosity Decipher genetic diversity in cattle breed dwelling in the colder regions of the world. Cytogenet. Genome Res. 164 (3-4), 154–164 (2024).
Boichard, D., Ducrocq, V., Croiseau, P. & Fritz, S. Genomic selection in domestic animals: principles, applications and perspectives. CR Biol. 339 (7–8), 274–277. https://doi.org/10.1016/j.crvi.2016 (2016). 04.007.
Jolliffe, I. T. & Cadima, J. Principal component analysis: a review and recent developments. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 374 (2065), 20150202. https://doi.org/10.1098/rsta.2015.0202 (2016).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based Estimation of ancestry in unrelated individuals. Genome Res. 19 (9), 1655–1664 (2009).
Sankararaman, S., Sridhar, S., Kimmel, G. & Halperin, E. Estimating local ancestry in admixed populations. Am. J. Hum. Genet. 82 (2), 290–303 (2008).
Reimand, J. et al. G: Profiler—a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 44 (W1), W83–W89 (2016).
NAGRP. Bioinformatics Coordination Program. Animal QTLdb. NAGRP Web site (2019). https://www.animalgenome.org/cgi-bin/QTLdb/index.
Pal, D. et al. Unraveling genetic admixture in the Indian crossbred cattle by different approaches using Bovine 50K BeadChip. Tropi. Anim. Health Prod. 54(2), 135 (2022).
Akanno, E. C. et al. Genomic prediction of breed composition and heterosis effects in Angus,Charolais, and Hereford crosses using 50K genotypes. Can. J. Anim. Sci. 97 (3), 431–438 (2017)..
Edwards, C. J., Loftus, R. T., Bradley, D. G., Dolf, G. & Looft, C. Relationships between the endangered Pustertaler–Sprinzen and three related European cattle breeds as analysed with 20 microsatellite loci. Anim. Genet. 31 (5), 329–332 (2000).
De Beukelaer, H., Badke, Y., Fack, V. & De Meyer, G. Moving beyond managing realized genomic relationship in long-term genomic selection. Genetics 206 (2), 1127–1138 (2017).
VanRaden, P. M. & Cooper, T. A. Genomic evaluations and breed composition for crossbred US dairy cattle. Interbull Bull. 2015, 49 (2015).
Goli, R. C. et al. Runs of homozygosity assessment using reduced representation sequencing highlight the evidence of random mating in emu (Dromaius novaehollandiae). Genome 68, 1–8 (2024).
FAO. FAOSTAT. Food and Agriculture Organization of the United Nations, Rome, Italy (2015).
Ahmad, S. F. et al. Population structure and admixture analysis in Frieswal crossbred cattle of India–a pilot study. Anim. Biotechnol. 31(1), 86–92 (2020).
Frkonja, A., Gredler, B., Schnyder, U., Curik, I. & Sölkner, J. Prediction of breed composition in an admixed cattle population. Anim. Genet. 43 (6), 696–703 (2012).
Edea, Z. et al. Genome-wide genetic diversity, population structure and admixture analysis in African and Asian cattle breeds. Animal 9 (2), 218–226 (2015).
Makina, S. O., Muchadeyi, F. C., van Marle-Köster, E., MacNeil, M. D. & Maiwashe, A. Genetic diversity and population structure among six cattle breeds in South Africa using a whole genome SNP panel. Front. Genet. 5, 333 (2014).
Nayee, N. et al. Suitability of existing commercial single nucleotide polymorphism chips for genomic studies in Bos indicus cattle breeds and their Bos taurus crosses. J. Anim. Breed. Genet. 135 (6), 432–441. https://doi.org/10.1111/jbg.12356 (2018).
Yang, S. et al. Parallel comparative proteomics and phosphoproteomics reveal that cattle myostatin regulates phosphorylation of key enzymes in glycogen metabolism and Glycolysis pathway. Oncotarget 9 (13), 11352 (2018).
Keating, N. & Quinlan, L. R. Small conductance potassium channels drive ATP-activated chloride secretion in the oviduct. Am. J. Physiol.-Cell Physiol. 302 (1), C100–C109 (2012).
Xia, J. et al. Genome-wide association study identifies loci and candidate genes for meat quality traits in Simmental beef cattle. Mammalian Genome 27, 246–255 (2016).
Rahman, J. U. et al. Genome-wide association studies of milk composition traits in indicine Badri cattle using DdRAD sequencing approach. Trop. Anim. Health Prod. 57 (1), 1–13 (2025).
Huang, C. et al. Whole-transcriptome analysis of longissimus dorsi muscle in cattle-yaks reveals the regulatory functions of ADAMTS6 gene in myoblasts. Int. J. Biol. Macromol. 262, 129985 (2024).
Congiu, M., Cesarani, A., Falchi, L., Macciotta, N. P. P. & Dimauro, C. Combined use of univariate and multivariate approaches to detect selection signatures associated with milk or meat production in cattle. Genes 15 (12), 1516 (2024).
Cánovas, A. et al. Multi-tissue omics analyses reveal molecular regulatory networks for puberty in composite beef cattle. PloS one 9 (7), e102551 (2014).
Ehrlich, K. C., Lacey, M. & Ehrlich, M. Epigenetics of skeletal muscle-associated genes in the ASB, LRRC, TMEM, and OSBPL gene families. Epigenomes 4, 1. https://doi.org/10.3390/epigenomes4010001 (2020).
Hooikaas, P. J. et al. MAP7 family proteins regulate kinesin-1 recruitment and activation. J. Cell Biol. 218 (4), 1298–1318 (2019).
Zaremba-Niedzwiedzka, K. et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541 (7637), 353–358 (2017).
Bao, H. et al. Platelet-derived extracellular vesicles increase Col8a1 secretion and vascular stiffness in intimal injury. Front. Cell Dev. Biol. 9, 641763 (2021).
Cai, Y., Yang, Y., Shen, M. & Zhou, T. Inhibition of cytokinesis by overexpression of nudcl that is localized to the centrosome and midbody. Cell Res. 19 (11), 1305–1308 (2009).
Tahir, M. S. et al. Meta-analysis of heifer traits identified reproductive pathways in Bos indicus cattle. Genes 12 (5), 768 (2021).
Duan, J. et al. Analysis of mRNA abundance for histone variants, histone-and DNA-modifiers in bovine in vivo and in vitro oocytes and embryos. Sci. Rep. 9 (1), 1217 (2019).
Doyle, J. L. et al. Genomic regions associated with skeletal type traits in beef and dairy cattle are common to regions associated with carcass traits, feed intake and calving difficulty. Front. Genet.. https://doi.org/10.3389/fgene.2020.00020 (2020).
Wu, S., Li, R. W., Li, W. & Li, C. J. Transcriptome characterization by RNA-seq unravels the mechanisms of butyrate-induced epigenomic regulation in bovine cells. PloS One 7 (5), e36940 (2012).
Strucken, E. M. et al. Genetic diversity and breed proportions of Indian stud cattle. In Proceedings of the World Congress on Genetics Applied to Livestock Production (WCGALP), Auckland, New Zealand (2018).
Dixit, S. P. et al. Genome analyses revealed genetic admixture and selection signatures in Bos indicus. Sci. Rep., 11 (1), 21924 (2021).
Singh, C. V. Cross-breeding in cattle for milk production: achievements, challenges and opportunities in India-A review. Adv. Dairy. Res. 4 (3), 158 (2016).
Taneja, V. K., Bhat, P. N. & Garg, R. C. Genetic divergence in various Sahiwal X Holstein crossbred grades. Theor. Appl. Genet. 54, 69–74 (1979).
Chen, Q. et al. Whole-genome resequencing reveals diversity, global and local ancestry proportions in yunling cattle.J. Anim. Breedi. Genet. 137 (6), 641–650 (2020).
Ward, J. A. et al. Genome-wide local ancestry and evidence for mitonuclear coadaptation in African hybrid cattle populations. Iscience 25, 7 (2022).
Khayatzadeh, N., Mészáros, G., Utsunomiya, Y. T. & Garcia, J. F. Locus-specific ancestry to detect recent response to selection in admixed Swiss Fleckvieh cattle. Anim. Genet. 47, 637–646. https://doi.org/10.1111/age.12470 (2016).
Pitt, D., Bruford, M. W., Barbato, M. & Orozco-terWengel, P. Demography and rapid local adaptation shape Creole cattle genome diversity in the tropics. Evol. Appl. 12, 105–122. https://doi.org/10.1111/eva.12641 (2018).
Yougbaré, B. et al. Local ancestry to identify selection in response to trypanosome infection in Baoulé x zebu crossbred cattle in Burkina Faso. Front. Genet. 12, 670390 (2021).
Settles, M. et al. A whole genome association analysis identifies loci associated with Mycobacterium avium subsp. paratuberculosis infection status in US holstein cattle.Anim. Genet. 40 (5), 655–662 (2009).
Miao, L. et al. SPOCK1 is a novel transforming growth factor-β target gene that regulates lung cancer cell epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 440 (4), 792–797 (2013).
Noyes, H., Brass, A., Obara, I. & Anderson, S. Genetic and expression analysis of cattle identifies candidate genes in pathways responding to Trypanosoma congolense infection. PNAS 108, 9304–9309. https://doi.org/10.1073/pnas.1013486108 (2011).
Kumar, M. A. N. O. J., Dahiya, S. P. & Ratwan, P. Current status and strategies for conservation of Hariana cattle. Indian J. Anim. Sci. 89 (6), 599–606 (2019).
Al Kalaldeh, M. et al. Detection of genomic regions that differentiate Bos indicus from Bos taurus ancestral breeds for milk yield in Indian crossbred cows. Front. Genet. 13, 1082802 (2023).
Yen, C. L., Stone, S. J., Koliwad, S., Harris, C. & Farese, R. V. Jr. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 49, 2283–2301. https://doi.org/10.1194/jlr.R800018-JLR200 (2008).
Grisart, B. et al. Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 12 (2), 222–231 (2002).
Sigdel, A., Abdollahi-Arpanahi, R., Aguilar, I. & Peñagaricano, F. Whole genome mapping reveals novel genes and pathways involved in milk production under heat stress in US Holstein cows. Front. Genet. 10, 473150 (2019).
Calderwood, S. K. et al. (2010). Signal transduction pathways leading to heat shock transcription.Signal Transduction insights 2, STI-S3994.
Jarolim, S. et al. Saccharomyces cerevisiae genes involved in survival of heat shock. G3 (Bethesda) 3, 2321–2333. https://doi.org/10.1534/g3.113.007971 (2013).
Saredi, G. et al. H4K20me0 marks post-replicative chromatin and recruits the TONSL-MMS22L DNA repair complex. Nature 534, 714–718. https://doi.org/10.1038/nature18312 (2016).
Dash, S., Singh, A., Dixit, S. P. & Kumar, A. Identification of selection signatures for milk performance traits among Indigenous dairy cattle breeds using high density genomic information. Indian J. Anim. Research 1, 7 (2022).
Chhotaray, S. et al. Genome-wide association study reveals genes crucial for coat color production in Vrindavani cattle. Livest. Sci. 247, 104476 (2021).
Reinsch, N. et al. A QTL for the degree of spotting in cattle shows synteny with the KIT locus on chromosome 6. J. Heredity 90 (6), 629–634 (1999).
Mészáros, G., Petautschnig, E., Schwarzenbacher, H. & Sölkner, J. Genomic regions influencing coat color saturation and facial markings in Fleckvieh cattle. Anim. Genet. 46 (1), 65–68 (2015).
Hayes, B. J., Pryce, J., Chamberlain, A. J., Bowman, P. J. & Goddard, M. E. Genetic architecture of complex traits and accuracy of genomic prediction: coat colour, milk-fat percentage, and type in Holstein cattle as contrasting model traits. PLoS Genet., 6 (9), e1001139 (2010).
Besmer, P. et al. The kit-ligand (steel factor) and its receptor c-kit/W: pleiotropic roles in gametogenesis and melanogenesis. Development 119 (Supplement), 125–137 (1993).
Koch, C. T., Bruggmann, R., Tetens, J. & Drögemüller, C. A non-coding genomic duplication at the HMX1 locus is associated with crop ears in Highland cattle. PLoS One 8(10), e77841. (2013).
Klawatsch, J. et al. Genetic basis of ear length in sheep breeds sampled across the region from the Middle East to the Alps. Animal Genet. 55 (1), 123–133 (2024).
Hu, M. et al. Assessing genomic diversity and signatures of selection in Chinese red steppe cattle using high-density SNP Array. Animals 13 (10), 1717 (2023).
Xiong, X. et al. RNA sequencing of the pituitary Gland and Association Analyses Reveal PRKG2 as a candidate gene for growth and Carcass Traits in Chinese Ningdu Yellow Chickens. Front. Vet. Sci. 9, 892024 (2022).
Bonnet, C. et al. Microdeletion at chromosome 4q21 defines a new emerging syndrome with marked growth restriction, mental retardation and absent or severely delayed speech. J. Med. Genet. 47 (6), 377–384 (2010).
Gangwar, M. et al. Identification of genetic variants affecting reproduction traits in Vrindavani cattle. Mamm. Genome 35 (1), 99–111 (2024).
Dado-Senn, B. et al. RNA-Seq reveals novel genes and pathways involved in bovine mammary involution during the dry period and under environmental heat stress. Sci. Rep. 8 (1), 11096 (2018).
de Lemos, M. V. A. et al. Association study between copy number variation and beef fatty acid profile of Nellore cattle. J. Appl. Genet. 59, 203–223 (2018).
Seo, M. et al. RNA-seq analysis for detecting quantitative Trait-Associated genes. Sci. Rep. 6, 24375. https://doi.org/10.1038/srep24375 (2016).
Tyagi, S. K. et al. Comparative signatures of selection analyses identify loci under positive selection in the Murrah Buffalo of India. Front. Genet. 12, 673697 (2021).
Yang, S. H. et al. Validation of PDE9A gene identified in GWAS showing strong association with milk production traits in Chinese Holstein. Int. J. Mol. Sci. 16, 26530–26542. https://doi.org/10.3390/ijms161125976 (2015).
Muramatsu, M. & Inoue, S. Estrogen receptors: how do they control reproductive and nonreproductive functions? Biochem. Biophys. Res. Commun. 270 (1), 1–10 (2000).
Utomo, B., Putranto, E. D. & Fadholly, A. Profile of follicle-stimulating hormone and polymorphism of follicle-stimulating hormone receptor in Madrasin cattle with ovarian hypofunction. Veterinary World 13 (5), 879 (2020).
Wang, Z. et al. Genome-wide scan identifies selection signatures in Chinese Wagyu cattle using a high-density SNP array. Animals 9 (6), 296 (2019).
Kumar, R. et al. Molecular characterization and polymorphism detection in HSPB6 gene in Sahiwal cattle. Indian J. Anim. Res. 49 (5), 595–598 (2015).
Makina, S. O. et al. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Selection Evol. 47, 1–14 (2015).
Bai, J. et al. iTRAQ-Based proteomic analysis reveals potential regulatory networks in dust mite-related asthma treated with subcutaneous allergen immunotherapy. Mol. Med. Rep. 22 (5), 3607–3620 (2020).
Yang, Q. et al. Genome-wide identification and comprehensive analyses of NAC transcription factor gene family and expression analysis under Fusarium kyushuense and drought stress conditions in Passiflora edulis. Front. Plant Sci. 13, 972734 (2022).
Ding, Y. et al. Bioinformatics analysis of LncRNA associated CeRNA network in melanoma. J. Cancer. 12 (10), 2921 (2021).
Khan, D. et al. Comparative physiological and transcriptomics profiling provides integrated insight into melatonin mediated salt and copper stress tolerance in Selenicereus undatus L.Plants 13 (24), 3602 (2024).
Acknowledgements
Authors would like to thank Director of ICAR-NDRI, ICAR-IIAB and CSB- CTRTI for providing a research facility to carry out this work and without their contributions, this work would not have been possible.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
RCG, KM, KKK: conceived and designed the experiments. KK and NS: performed the experiments. KC, SC, PR, CSC, OSS and VD: analyzed the data. RCG, KM, NS, KKK, AK, GNA: wrote the paper. KC, SC, PR, CSC, IG, SS and SPD: edited the paper. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Goli, R.C., Chishi, K.G., Mahar, K. et al. Genome wide locus-specific ancestry analysis revealed adaptive admixtures in crossbred cattle of India. Sci Rep 15, 17069 (2025). https://doi.org/10.1038/s41598-025-01971-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-01971-7
