
Abstract
In this study, cetylpyridinium peroxyphosphotungstate was synthesized as an environmentally benign phase transfer catalyst with high catalytic activity from cetylpyridinium chloride, phosphotungstic acid, and hydrogen peroxide. The chemical structure of the catalyst was identified by SEM, XRD, FT-IR, ICP, elemental analysis, XPS, and TGA/DTG, revealing its catalytic activity to be related to the regular and dense distribution of pores on its surface. High catalytic activity was demonstrated in the oxidation of linalool to furanoid and pyranoid linalool oxides using hydrogen peroxide in low toxicity organic solvents such as ethanol and ethyl acetate. The total yield and total selectivity of furanoid and pyranoid linalool oxides were optimized by varying parameters including the catalyst loading, hydrogen peroxide amount, reaction temperature, and reaction time. Optimized conditions providing furanoid and pyranoid linalool oxides with 80.1% total yield were 10 wt% catalyst and 1.16 mL/g 30% H2O2 (vs. linalool) at 30 °C for 1.5 h. This procedure has the advantages of high yield, employing a green oxidant, and non-toxic reagents.
Similar content being viewed by others
Introduction
Phosphotungstic acid (H3PW12O40), after treatment with hydrogen peroxide, has high catalytic performance in the oxidation of olefinic C = C, and is commercially available and stable. It is considered environmentally benign, giving clean reaction processes,1,2 and is a highly active solid acid with potential as an alternative to traditional metal catalysts3,4 Various oxidants such as peracetic acid, NaOCl, and tert-butyl hydroperoxide have been used to selectively oxidize olefins. However, these oxidants form reduced byproducts,5 while the use of hydrogen peroxide as an oxidant is ideal due to its high oxygen atom efficiency with co-production of water6,7 Moreover, hydrogen peroxide requires no extra acid for olefin oxidation, aligning the process with the concept of environmentally friendly chemistry8 Phosphotungstic acid is only suitable for homogeneous reaction mixtures. However, the oxidation of C = C usually requires reaction under heterogeneous conditions, especially when hydrogen peroxide is used as the oxidant. To be a viable oxidant in heterogeneous systems, phosphotungstic acid can be supported on a less polar carbon chain to construct a phase transfer catalyst.
Alkyl pyridinium chloride belonging to quaternary ammonium salts, is an excellent class of surfactants. The quaternary ammonium cation of these compounds could combine with the phosphotungstic acid anion through ionic bonding to form alkylpyridine phosphotungstate, rather than physically adsorbing the phosphotungstic acid ion. This coupling configuration effectively inhibits the leaching of phosphotungstic acid anions during repeated use of alkyl pyridinium chloride, consequently ensuring good reusability performance. Therefore, alkylpyridine phosphotungstate is considered to be a good phase transfer catalyst. In a heterogeneous reaction system composed of aqueous and organic phases using hydrogen peroxide as an oxidant, the counter cation of alkylpyridine phosphotungstate could bind to hydrogen peroxide, forming a high concentration reaction microenvironment on the catalyst surface and accelerating the reaction process9.
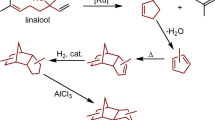
Linalool is an important natural product belonging to the chain terpenoids10 Furanoid linalool oxides and pyranoid linalool oxides are widely used in food, medicine, and other fields due to their bacteriostatic, insecticidal, and other biological activities, and their market potential is huge11,12,13,14,15,16,17 Hence, oxidation of linalool has received extensive research attention. At present, there are two main methods to prepare furanoid or pyranoid linalool oxides from linalool. In the first, linalool is oxidized with hydrogen peroxide in the presence of an organic acid catalyst (Fig. 1)18,19 The reaction conditions of this method are mild and the reaction steps are simple. However, the post-treatment is troublesome and the organic acids used can corrode the equipment and cause serious environmental pollution. In the second method, linalool oxide is provided from linalool analogs under enzyme catalysis (Fig. 2)20,21 Specific isomers of unsaturated alcohols can be obtained by this method but it requires a multi-step reaction, product yields tend to be low, and the enzymes and raw materials are difficult to obtain. Therefore, the preparation technology for furanoid and pyranoid linalool oxides still needs to be optimized.

Synthesis of linalool oxide with H2O2 and organic acid catalysts.

Synthesis of linalool oxide with enzyme catalysts.
This study describes the synthesis of cetylpyridinium peroxyphosphotungstate (an alkylpyridine phosphotungstate) (Fig. 3) and its use as a catalyst for the preparation of furanoid and pyranoid linalool oxides from linalool with H2O2 oxidant. The results of the study also have positive significance for the development of value-added processing products of linalool.

Synthesis of cetylpyridinium peroxyphosphotungstate.
Experimental
Materials
All reagents were purchased from Shanghai Jingchun Biochemical Technology Co., Ltd. These reagents were of analytical grade and used without further purification.
The Preparation of cetylpyridinium peroxyphosphotungstate [π-C5H5NC16H33]3PW4O24
A modified literature procedure22,23,24,25 was applied. Cetylpyridinium chloride (2.62 g, 7.7 mmol) and 30% H2O2 (84 mL) were stirred at 40 ºC in a 250 mL 3-necked flask with a thermometer, a reflux condenser and a constant pressure funnel. To the flask was added dropwise a solution of phosphotungstic acid (7.20 g, 2.5 mmol) in 30% H2O2 (21 mL) through the constant pressure funnel and the reaction temperature was maintained at 40 ºC for 5 h. After completion of the reaction, decompressional filtration, washing, and air drying, cetylpyridinium peroxyphosphotungstate (3.71 g, 1.8 mmol, 72.0% based on P) was obtained as a light yellow solid.
Characterization of cetylpyridinium peroxyphosphotungstate
To study the surface morphological of the sample, SEM micrographs were recorded on a S-570 scanning electron microscope (Hitachi, Japan). XRD analysis of the sample was carried out using a D/MAX 2500 V diffractometer (Rigaku, Japan) with Ni-filtered Cu Kα radiation (λ = 0.154 nm). Powder X-ray difraction patterns were measured in the range of 5° to 90° with a scanning rate of 2 °/min. Analyses were performed at a voltage of 40 kV and a current of 40 mA. A Thermo Nicolte IS10 spectrometer (Thermo Fisher Scientific, USA) was employed to carried out the Fourier transform infrared (FT-IR) spectra of the synthesized catalyst. The content of P and W in the sample was analysed by a PerkinElmer 2100 inductively coupled optical emission spectrometer (ICP-OES). The element analysis was recorded on a Vario EL cube (Elemantar, Germany). X-ray photoelectron spectroscopy (XPS) was utilized to study the surface chemistry of the sample with a Thermo Scientific Nexsa instrument from Thermo Scientific (Thermo Fisher Scientific, Waltham, USA), with a monochromated Al Kα source (1486.6 eV), at a base pressure < 1 × 10− 9 mbar. The pass energies for the survey spectra and high-resolution spectra were 200 eV and 20 eV, respectively. The deconvolution of C1s, O1s, and W4f spectra were carried out using a Gaussian-Lorentzian function. A simultaneous analysis of thermogravimetry (TGA) and derivative thermal gravimetry (DTG) was executed by a Discovery TGA 55 thermogravimetric analyzer ( TA Instruments, USA).
Oxidation of Linalool to furanoid and pyranoid Linalool oxides with cetylpyridinium peroxyphosphotungstate catalyst
Linalool (8.16 mL, 49.16 mmol), cetylpyridinium peroxyphosphotungstate (0.8 g), an organic solvent such as ethanol, ethyl acetate, or trichloromethane (20 mL) and 30% H2O2 (13.2 mL) were added to a 100 mL flask with a thermometer and a reflux condenser. The reaction mixture was stirred for 1.5 h at 30 ºC. After completion of the reaction, the mixture was filtered by vacuum suction and the filtrate was dried over anhydrous sodium sulfate. GC-MS was performed to determine the yields of furanoid linalool oxides and pyranoid linalool oxides (peak area normalization method) and their chemical structures.
The process total yield and selectivity for furanoid linalool oxides and pyranoid linalool oxides were optimized in experiments varying the solvent, reaction time, reaction temperature, catalyst loading, and the amount of 30% H2O2. Each test was repeated three times.
GC analysis
The yields of furanoid linalool oxides and pyranoid linalool oxides were measured on a GC-2014 AF (Shimadzu, Kyoto, Japan) equipped with a HP-5 quartz capillary column (30 m × 0.25 mm i.d. × 0.25 µm flm thickness). The separation conditions gas chromatographic were as follows: FID detector; carrier gas nitrogen; flow rate 3.0 ml/min; injection temperature 270 °C; detector temperature 270 °C. The initial column temperature was kept at 60 °C for 0.5 min; programmed to 80 °C at a rate of 5 °C/min and kept constant at 80 °C for 0.5 min; programmed to 140 °C at a rate of 20 °C/min and kept constant at 140 °C for 0.5 min; programmed to 180 °C at a rate of 4 °C/min and kept constant at 180 °C for 0.5 min; programmed to 230 °C at a rate of 10 °C/min and kept constant at 230 °C for 1 min; programmed to 280 °C at a rate of 10 °C/min and kept constant at 280 °C for 1 min.
GC-MS analysis
The chemical structures of furanoid linalool oxides and pyranoid linalool oxides were identified by GC-MS. GC-MS system was composed of an Agilent Technologies 7890 A gas chromatograph and an Agilent 7683B auto injector equipped with a 5975 C VL Agilent mass selective detector (Agilent Technologies Inc., USA). The scan rate of the mass spectral was 2.85/s and the scan range was 50–500 Da in splitless injection mode with a helium flow rate of 0.7 ml/min. The column head pressure was maintained at 10 psi. The MS detector was operated in the EI mode with an ionization voltage of 70 eV and a source temperature of 230 °C. The temperature of the GC transfer line and injector were 300 °C and 280 °C, respectively. Chromatographic separation was carried out on a DB-5MS capillary column (30 m × 0.25 mm i.d. × 0.25 µm flm thickness). The temperature procedure has been described in the previous section.
Results and discussion
Characterization of cetylpyridinium peroxyphosphotungstate
The structure of cetylpyridinium peroxyphosphotungstate was characterized by SEM, XRD, FT-IR, ICP, element analysis, XPS, and TGA/DTG.
The surface morphology of cetylpyridinium peroxyphosphotungstate was observed by SEM (Fig. 4a and b), revealing the uniform and densely distributed pores on the catalyst surface that increase the specific surface area, enhancing reactant adsorption and reaction site density. As a result, the catalytic reaction rate could be increased. To study the distribution and chemical composition of the catalyst, EDX analysis was performed (Fig. 4c). The elemental peaks attributed to carbon, oxygen, tungsten, and phosphorus in the EDX spectrum confirm the presence of these elements in the obtained catalyst. In comparison with phosphotungstic acid (H3PW12O40), the W/P atomic ratio of the catalyst was found to decrease from 12:1 to 4:1. This clearly indicates that the valence state of tungsten changes after phosphotungstic acid is supported on cetylpyridinium chloride under the action of hydrogen peroxide.

(a and b) SEM images and (c) EDS results of cetylpyridinium peroxyphosphotungstate.
The XRD diffraction pattern was obtained by processing the XRD raw data with Origin 8. The XRD diffractogram of cetylpyridinium peroxyphosphotungstate (a) is illustrated in Fig. 5. For comparison, the diffractogram of phosphotungstic acid (b) is also presented in Fig. 5. In the XRD pattern of phosphotungstic acid, diffraction peaks at 2θ = 6.8°, 8.5°, 10.3°, 18.4°, 25.2°, and 34.9° originate from the (010), (200), (102), (410), (133), and (525) diffraction facets, respectively, of H3PW12O40 (JCPDS File No. 50–0655). This is consistent with the characteristic diffraction peaks of Keggin phosphotungstate reported in the literature26,27 Moreover, the crystallinities of all the above crystalline phases were over 95%. In contrast, in the XRD pattern of cetylpyridinium peroxyphosphotungstate, a broad amorphous hump was found at 2θ = 20°-30°, suggesting the dominance of disordered structures. Strong diffraction peaks of the catalyst are only presented at 2θ = 8.3°, 8.7°, and 8.9°, and all these crystal phases have crystallinities below 90%. Compared with phosphotungstic acid, the crystallinity of each crystal phase of cetylpyridinium peroxyphosphotungstate is significantly reduced, indicating that the introduction of counter cations and changes in tungsten valence state have a significant impact on its crystal structure.

XRD patterns of cetylpyridinium peroxyphosphotungstate (a) and phosphotungstic acid (H3PW12O40) (b).
The FT-IR spectra of cetylpyridinium chloride, phosphotungstic acid, and cetylpyridinium peroxyphosphotungstate were shown in Fig. 6. In the FT-IR spectrum of cetylpyridinium chloride, bands at 1639, 1578, and 1485 cm− 1 correspond to the stretching vibration of C = C, the stretching vibration of C = N+, and the bending vibration of C − H, respectively. The spectrum of cetylpyridinium peroxyphosphotungstate also shows these characteristic bands at 1632, 1578, and 1486 cm− 1, consistent with those of cetylpyridine chloride. However, based on the references ,8,28 it could be inferred that five new bands at 1080, 951, 893, 821, and 545 cm− 1 correspond to the stretching vibration of P − O, and W = O, W − O−W, O − O, and W − O−O, respectively, indicating that phosphotungstic acid was successfully supported on cetylpyridinium chloride. In the FT-IR spectrum of phosphotungstic acid, bands at 1081, 986, and 894 cm− 1 can be attributed to the stretching vibrations of P − O, W = O, and W − O−W. However, bands for the stretching vibrations of O − O and W − O−O are absent in the spectrum of phosphotungstic acid. Hence, it can be concluded that phosphotungstic acid reacts under the action of hydrogen peroxide to produce peroxide bonds in cetylpyridinium peroxyphosphotungstate.

FT-IR spectra of cetylpyridinium chloride, phosphotungstic acid, and cetylpyridinium peroxyphosphotungstate.
To confirm the elemental composition of cetylpyridinium peroxyphosphotungstate, inductively coupled plasma (ICP) analysis was used to determine the W and P contents. The synthesis of quaternary ammonium peroxyphosphotungstate with phosphotungstic acid and quaternary ammonium salts with H2O2 as oxidizing agent has been reported, and the elemental composition of the anion {PO4[WO(O2)2]4}3− has been identified28,29 In our cetylpyridinium peroxyphosphotungstate catalyst, the contents of W and P were 35.28 wt% and 1.49 wt%, respectively, representing a W to P molar ratio of 4:1, in agreement with the literature8,28 In addition, elemental analysis revealed the C, H, and N contents to be 36.75, 5.51, and 2.12 wt%, respectively. From these combined data, we can reasonably conclude that the molar ratio of C, H, N, P, W, and O is 63 : 114 : 3 : 1: 4 : 24.
XPS analysis was performed to measure the change in the composition and chemical state of the elements on the surface of the samples. In this study, avantage software (Thermo Fisher Scientific, Waltham, USA) was used for analyzing XPS data (Fig. 7). As a result, it was found that the survey spectra of cetylpyridinium chloride show the four characteristic peaks O1s, N1s, C1s, and Cl2p centered at 532.05, 401.86, 285.05, and 197.18 eV. The XPS survey of cetylpyridinium peroxyphosphotungstate found five characteristic peaks at 531.25, 401.93, 285.02, 133.26, and 36.15 eV corresponding to O1s, N1s, C1s, P2p, and W4f signals, respectively. The presence of P2p and W4f further confirmed that phosphotungstic acid was successfully supported on cetylpyridinium chloride. In the XPS survey of phosphotungstic acid, the three peaks at 531.28, 134.80, and 37.13 eV can be attributed to O1s, P2p, and W4f, respectively. After chemical modification of phosphotungstic acid, the binding energy of P2p and W4f decreased. The change in O/W ratio between phosphotungstic acid (3.41:1) and cetylpyridinium peroxyphosphotungstate (4.68:1) is significant and indicates that the chemical state of tungsten element changed when phosphotungstic acid was grafted onto cetylpyridinium chloride.

Survey XPS spectra of cetylpyridinium chloride, phosphotungstic acid, and cetylpyridinium peroxyphosphotungstate.
High resolution C1s, O1s, and W4f spectra were also analyzed for a better understanding of the surface chemistry (Figs. 8, 9 and 10). After deconvolution, two sub-peaks for the C1s spectra of cetylpyridinium chloride situated at 284.69 and 285.23 eV can be attributed to the C = C (C1s-1) and C − C/C − H (C1s-2) bonds, respectively. The C1s spectra of cetylpyridinium peroxy- phosphotungstate also presented two sub-peaks situated at 284.28 (C1s-1) and 285.67 eV (C1s-2). After cetylpyridinium chloride modification, the relative content of C1s-1 decreased from 50.22 to 34.73% and that of C1s-2 increased from 49.78 to 65.27% (relative content = C1s-1 or C1s-2 content / total C1s content × 100%). Therefore, more carbon chains (hexadecyl) and fewer pyridine rings were exposed on the surface of cetylpyridinium chloride.

High resolution C1s spectra of cetylpyridinium chloride and cetylpyridinium peroxyphosphotungstate.

High resolution O1s spectra of phosphotungstic acid and cetylpyridinium peroxyphosphotungstate.

High resolution W4f spectra of phosphotungstic acid and cetylpyridinium peroxyphosphotungstate.
The high resolution O1s spectra of phosphotungstic acid revealed two distinct peaks at 530.95 (O1s-1) and 531.83 eV (O1s-2), attributed to the P − O−H (O1s-1) and P = O/W = O (O1s-2) bonds, respectively. In contrast, the O1s spectra of cetylpyridinium peroxyphosphotungstate present three distinct peaks at 529.85 eV (lattice oxygen), 531.32 eV (surface oxygen species), and 532.23 eV (P − O−W bond). Surface oxygen in the catalyst can be classified as W(O)2, and W = O can be attributed to highly oxidized oxygen30 It should be noted that surface oxygen is involved in the catalytic reaction and could be beneficial for the formation of various oxygen-containing species during the catalytic process8,31.
The high resolution W4f spectra of phosphotungstic acid present two distinct peaks, W4f7/2 at 35.94 and W4f5/2 at 38.09 eV, attributed to W6+. However, the high resolution W4f spectra of cetylpyridinium peroxyphosphotungstate revealed four distinct peaks. The peaks of W4f7/2 at 34.81 eV and W4f5/2 at 37.0 eV are attributed to W4+, while those of W4f7/2 at 35.18 eV and W4f5/2 at 38.61 eV indicate W5+. This result suggests that the valence state of most surface W atoms in the cetylpyridinium peroxyphosphotungstate catalyst decreased from + 6 to + 5/+4, signifying that lower tungsten valence states are predominant in the catalyst.
The TGA-DTG analysis of cetylpyridinium peroxyphosphotungstate during pyrolysis in a N2 atmosphere is illustrated in Fig. 11. The TGA curve of cetylpyridinium peroxyphosphotungstate exhibits several distinct peaks, indicating several notable weight loss processes. The first weight loss is observed between room temperature and 345 ºC and can be mainly attributed to the elimination of a small amount of adsorbed and crystallized water. The second major weight loss between 345 and 405 ºC could be ascribed to the decomposition of the alkyl chain. When the temperature reaches 405 ºC, the peroxophosphotungstate ion in the catalyst begins to decompose gradually.

TGA-DTG curve of cetylpyridinium peroxyphosphotungstate.
Oxidation of Linalool to furanoid and pyranoid Linalool oxides with cetylpyridinium peroxyphosphotungstate catalyst
To date, most described approaches for synthesis of furanoid and pyranoid linalool oxides are based on the epoxidation of linalool followed by cyclization under acid-catalysis,15 usually with peroxy organic acids, with yields over 60%. However, peroxy organic acids are associated with high risk and toxicity. Moreover, green synthesis protocols are becoming increasingly important12 In this context, due to the environmentally benign nature of H2O2 and the high catalytic performance of phosphotungstic acid, research into the oxidation of linalool with these reagents has aroused great interest. Nevertheless, in our previous investigation, furanoid and pyranoid linalool oxides could not be synthesized from linalool using H2O2 and phosphotungstic acid due to limited contact between reactant and reagents under these heterogeneous conditions. To resolve this issue, we designed and synthesized cetylpyridinium peroxyphosphotungstate catalyst. Presenting both hydrophobic carbon chains and hydrophilic peroxophosphotungstate ions, it can behave as a phase transfer catalyst in heterogeneous conditions. In addition, the many pores on the catalyst surface could effectively improve the catalyst performance.
To our delight, linalool oxidation using cetylpyridinium peroxyphosphotungstate catalyst and H2O2 oxidant resulted in a high total yield of furanoid and pyranoid linalool oxides. According to GC analysis, five main products were formed in this reaction (Fig. 12) and their chemical structures were identified by GC-MS.

GC spectra of the linalool oxidation reaction mixture.
MSD chemstation data Analysis F.01.00 (Thermo Fisher Scientific, Waltham, USA) was used for analyzing GC-MS data. The obtained mass spectra of the products were retrieved using the NIST 11. L standard mass spectrometry library (National Institute of Standards and Technology, Gaithersburg, USA). As a result, it was found that the GC-MS spectrogram matching degrees for trans-furanoid linalool oxide (4a), cis-furanoid linalool oxide (4b), trans-pyranoid linalool oxide (5a), cis-pyranoid linalool oxide (5b), and 2,2,6-trimethyl-6-vinyldihydro-2 H- pyran-3(4 H)-one (6) were 96, 96, 95, 96 and 96%, respectively. The reaction mechanism is shown in Fig. 13. First, the 6-C = C bond of linalool is oxidized by hydrogen peroxide in the presence of the catalyst to provide epoxide (2), hydrolysis of which gives triol (3). Next, a molecule of water is eliminated by the interaction between the 6-OH and 3-OH of 3 to provide 4a and 4b. At the same time, 5a and 5b are obtained through the interaction between the 7-OH and 3-OH of 3 with elimination of a molecule of water. The 3-C = C bond of linalool was more difficult to oxidize by hydrogen peroxide than the 6-C = C bond, presumably due to the formation of secondary and tertiary carbon free radicals, respectively. The lower stability of the former results in easier oxidation of the 6-C = C bond.

The reaction route for linalool furanoid and pyranoid oxidation products.
The synthesis of furanoid and pyranoid linalool oxides was optimized by varying the solvent, reaction time, reaction temperature, the amount of 30% H2O2, and the catalyst loading as process parameters. The conversion, selectivity and yield were calculated according to Eq. (1), Eq. (2), and Eq. (3), respectively. IBM SPSS Statistic 20 was used for statistical analysis of the data. A one-way ANOVA and effect sizes were used to evaluate the effect of the process parameters on the conversion of linalool, the total yield of furanoid linalool oxides and pyranoid linalool oxides, and the total selectivity (Tables 1, 2, 3, 4, 5, 6, 7, 8, 9 and 10). Significant differences were observed for the effects of modifying reaction temperature, reaction time, the amount of 30% H2O2, and the amount of catalyst on linalool conversion (P < 0.01). Effect sizes for these factors on linalool conversion were higher than 0.85. Among them, the effect size of the amount of catalyst was the largest at 0.999. In addition, the influence of solvent, the amount of 30% H2O2, and the catalyst loading on the total yield and total selectivity for furanoid and pyranoid linalool oxides showed significant differences (P < 0.01) (Tables 2, 4, 6 and 8, and 10), and all the parameters showed high effect sizes of more than 0.88. However, the solvent had little effect on the conversion of linalool and no significant differences were observed for the effects of modifying reaction time and reaction temperature on the total yield and total selectivity.
The effect of solvent on the synthesis of furanoid and pyranoid linalool oxides is shown in Table 1. The linalool conversion could be 100% when ethanol, ethyl acetate, or trichloromethane was used as the solvent. Both the total yield and the total selectivity for furanoid and pyranoid linalool oxides were strongly affected by solvent. The highest yield (74.4%) and selectivity (74.4%) were obtained with ethanol as solvent. Therefore, ethanol was considered optimal.
The effect of reaction time on the synthesis of furanoid and pyranoid linalool oxides is detailed in Table 3. The highest yield (75.6%) and selectivity (75.6%) were provided after 1.5 h. Therefore, 1.5 h was considered optimal.
The effect of reaction temperature on the synthesis of furanoid and pyranoid linalool oxides is shown in Table 5. The total yield and total selectivity are seen to increase with increasing reaction temperature up to 30 ºC, while temperature had little effect in the range 30 − 60 ºC. Therefore, 30 °C was considered optimal.
The effect of the amount of 30% H2O2 on the synthesis of furanoid and pyranoid linalool oxides is detailed in Table 7. Both the total yield and the total selectivity were strongly affected by the amount of 30% H2O2. Using 3.3 mL 30% H2O2, the total selectivity could be increased to 95.3%, although the total yield was only 57.4% under this condition due to low conversion of linalool. The highest yield (80.1%) and conversion of linalool (99.3%) were provided using 6.6 mL 30% H2O2, but the total selectivity decreased to 80.6% due to increased by-product formation. On balance, the optimal amount of 30% H2O2 was considered to be 6.6 mL (1.16 g/mL H2O2 vs. linalool).
The effect of catalyst loading on the synthesis of furanoid and pyranoid linalool oxides is detailed in Table 9. The total yield and total selectivity could be increased with increasing catalyst loading in the range 0.4–0.8 g (5 − 10 wt% vs. linalool), though a further increase (> 0.8 g, > 10 wt% vs. linalool) had little effect. Therefore, the optimal catalyst loading was 0.8 g (10 wt%).
l1: amount of linalool before reaction; l2: amount of linalool after reaction.
x1: the theoretical total amount of furanoid and pyranoid linalool oxides is obtained when all linalool is converted to linalool oxides; x2: the actual total amount of furanoid and pyranoid linalool oxides is obtained after the reaction.
Conclusion
In summary, cetylpyridinium peroxyphosphotungstate has been prepared from cetylpyridinium chloride, H2O2, and phosphotungstic acid. The chemical structure of this material was characterized using SEM, XRD, FT-IR, ICP, elemental analysis, XPS, and TGA/DTG. The results of these analysis support the expected structure of cetylpyridinium peroxyphosphotungstate. As a catalyst for the oxidation of linalool with H2O2, cetylpyridinium peroxyphosphotungstate provides furanoid linalool oxides and pyranoid linalool oxides in high yield and selectivity in low toxicity organic solvents like ethanol and ethyl acetate. Optimum oxidation conditions were identified as 10 wt% catalyst and 1.16 g/ mLH2O2 (vs. linalool) at 30 °C for 1.5 h. Cetylpyridinium peroxyphosphotungstate catalyst has the advantages with strong catalytic activity, low toxicity, and low corrosiveness to equipment, and could be used as an efficient, green and environmentally friendly catalyst for catalytic oxidation of C = C. Most important of all, it will have a wide range of application scenarios in the green catalytic oxidation of C = C for synthesis of oxygen derivatives or adjacent diol compounds.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Reddy, K. M., Babu, N. S., Suryanarayana, I., Prasad, P. S. S. & Lingaiah, N. The silver salt of 12-tungstophosphoric acid: an efficient catalyst for the three-component coupling of an aldehyde, an amine and an alkyne. Tetrahedron Lett. 47, 7563–7566 (2006).
Firouzabadi, H., Iranpoor, N. & Amani, K. Solvent-free and selective oxidation of hydroxy groups to their corresponding carbonyl functions with ferric nitrate activated by heteropoly acids. Synthesis 3, 408–412 (2003).
Kozhevnikov, I. V. Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem. Rev. 98, 171–198 (1998).
Wang, G., Shen, Y., Wu, X. & Wang, L. Solvent-free hydroalkylation of olefins with 1,3-diketones catalyzed by phosphotungstic acid. Tetrahedron Lett. 49, 5090–5093 (2008).
Trost, B. M. The atom economy-a search for synthetic efficiency. Science 254, 1471–1477 (1991).
Kon, Y. et al. Continuous synthesis of epoxides from alkenes by hydrogen peroxide with titanium silicalite-1 catalyst using flow reactors. Adv. Synth. Catal. 365, 3227–3233 (2023).
Liu, M. & Zhang, G. Amorphous goethite as a catalyst of chemoselectivity epoxidation of alkenes by hydrogen peroxide. Russ J. Gen. Chem. 92, 1539–1545 (2022).
Zhang, L. Z. et al. Study on the epoxidation of chain olefins using biquaternary ammonium phosphotungstic acid phase transfer catalysts under no-solvent condition. Chem. Eur. J. 30, 1–11 (2024).
Ishii, Y. et al. Hydrogen peroxide oxidation catalyzed by heteropoly acids combined with cetylpyridinium chloride. Epoxidation of olefins and allylic alcohols, ketonization of alcohols and diols, and oxidative cleavage of 1, 2-diols and olefins. J. Org. Chem. 53, 3587–3593 (1988).
Krill, C., Rochfort, S. & Spangenberg, G. A. High-throughput method for the comprehensive analysis of terpenes and terpenoids in medicinal cannabis biomass. Metabolites 10, 276–283 (2020).
Majumder, S., Ghosh, A. & Bhattacharya, M. Natural anti-inflammatory terpenoids in Camellia japonica leaf and probable biosynthesis pathways of the metabolome. Bull. Natl. Res. Cent. 44, 2016–2023 (2020).
Wei, T. P. et al. Catalytic metal-free allylic C-H amination of terpenoids. J. Am. Chem. Soc. 142, 16716–16722 (2020).
Arsheed, A. et al. A novel terpenoid class for prevention and treatment of KRAS-driven cancers: comprehensive analysis using in situ, in vitro, and in vivo model systems. Mol. Carcinog. 59, 886–896 (2020).
Vincent, O. et al. Linalool oxide: generalist plant based lure for mosquito disease vectors. Parasites Vectors. 8, 581–590 (2015).
Negromonte, F. S. M. et al. Anxiolytic-like effects of inhaled Linalool oxide in experimental mouse anxiety models. Pharmacol. Biochem. Behav. 100, 259–263 (2011).
Negromonte, F. S. M. et al. Antinociceptive and anticonvulsant effects of the monoterpene Linalool oxide. Pharm. Biol. (Abingdon U K). 55, 63–67 (2017).
Satoh, M. et al. Antitermitic and antifungal properties of enantiopure Linalool and furanoid Linalool oxide confirmed in Lindera umbellata Var. Membranacea. J. Wood Chem. Technol. 42, 37–45 (2022).
Yang, H., Wen, S., Qiu, F., Zhang, T. & Wu, Y. Study on the influence of different catalytic systems on the synthesis of Linalool oxide. Appl. Chem. Ind. (Xi’an China). 48, 2570–2573 (2019).
Ma, J., Liu, H., Huang, C. & Mo, Y. Oxidation synthesis of phase transfer catalyist of Linalool oxide. J. Guangxi Teachers Educ. Univ. (Natural Sci. Edition). 5, 5–8 (2004).
Matthijs, J. V. T. et al. Enantioselective Bio-Hydrolysis of Geranyl-Derived rac-Epoxides: A chemoenzymatic route to trans-Furanoid Linalool oxide. Adv. Synth. Catal. 2, 813–825 (2019).
Serra, S., De Simeis, D. & Brenna, E. Lipase mediated resolution of cis- and trans-linalool oxide (pyranoid). J. Mol. Catal. B: Enzym. 3, S425 (2017).
Zhao, W., Ma, B., Hua, H., Zhang, Y. & Ding, Y. Environmentally friendly and highly efficient alkenes epoxidation system consisting of [π-C5H5N(CH2)11]3PW4O32 / H2O2 / Ethyl acetate / olefin. Catal. Commun. 9, 2455–2459 (2008).
Hua, A., Ma, B. C., Tong, D. J., Zhao, W. & Ding, Y. [π-C5H5N(CH2)11]3[PMoW3O24]: A heteropolyoxmolybdotungstate catalyst for efficient and recyclable epoxidation of 1-octene with 30% H2O2 using environment friendly solvent. J. Mol. Catal. 23, 98–105 (2009).
Zhao, X. et al. Epoxidation of allyl chloride with H2O2 catalyzed by three structurally related quaternary ammonium modified polyoxophosphotungst. Appl. Catal. A. 608, 1–7 (2020).
Huang, D. Z., Zhu, S. J., Lan, H. Y. & Lei, F. H. A novel method for Preparation of (3R, 4R)-4, 7, 7-trimethyl-6-oxabicyclo [3.2.1] octane-3, 4-diol. Chem. Ind. Prod. 35, 9–15 (2015).
Hu, C. W. et al. Synthesis of new types of polyoxometallate pillared anionic clays: 31p and 27Al MAS NMR study of the orientation of intercalated PW11VO404–. Chem. Commun. 2, 121–122 (1996).
Jiang, S. et al. Preparation of biodiesel catalyzed by phosphotungstic acid supported on amino functionalized silica and its kinetics. J. Chin. Cereals its Association. 39, 149–156 (2024).
Venturello, C. & D’Aloisio, R. Quaternary ammonium tetrakis(diperoxotungsto)phosphates (3-) as a new class of catalysts for efficient alkene epoxidation with hydrogen peroxide. J. Org. Chem. 53, 1553–1557 (1988).
Yadav, G. D. & Mistry, C. K. A new, effective catalytic system for epoxidation of olefins by hydrogen peroxide under phase-transfer conditions. J. Mol. Catal. A: Chem. 172, 135–149 (2001).
Wang, Y. Z., Li, Z. Z., Liu, Z. W. & Shi, X. Y. Heterogenous carboxyl-functionalized bilayer ionic liquids/polyoxometalate catalysts for extractant-free oxidative desulfurization. J. Mol. Liq. 373, 1–15 (2023).
Chen, H. et al. Activating lattice oxygen in perovskite oxide by B-site cation do for modulated stability and activity at elevated temperatures. Adv. Sci. 8, 1–13 (2021).
Acknowledgements
The authors acknowledge the financial support from the Characteristic Innovation Project of Guangdong Colleges and Universities in 2022 (grant No. 2022KTSCX075).
Funding
This research was financially supported by the Characteristic Innovation Project of Guangdong Colleges and Universities in 2022 (grant No. 2022KTSCX075), Innovation and Entrepreneurship Training Program for College Students in 2023 (2023012), the Project of Key Laboratory of General Universities in Guangdong Province (grant No. 2023KSYS007).
Author information
Authors and Affiliations
Contributions
S.Z. and C.Y. wrote the main manuscript text, J.Z. and R.Z. prepared Figs. 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12, and L.L. and X.H. prepared Tables 1, 2, 3, 4, 5, 6, 7, 8, 9 and 10. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, S., Chen, Y., Zhou, R. et al. Synthesis and characterization of cetylpyridinium peroxyphosphotungstate and their catalytic properties for linalool oxidation. Sci Rep 15, 18616 (2025). https://doi.org/10.1038/s41598-025-03851-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-03851-6
