
Abstract
In the pursuit of therapeutic agents for Alzheimer’s disease (AD), this study employed a molecular hybridization strategy to design and synthesize substituted cinnamoyl piperidinyl acetates. A total of 17 novel derivatives were evaluated for their inhibitory effects against acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), key enzymes implicated in AD pathogenesis. Notably, compound 5b, featuring a 2-chloro substitution, emerged as the most potent AChE inhibitor (IC50 = 19.74 ± 0.96 µM), while compound 5q, possessing a 4-ethoxy-3-methoxy moiety, demonstrated superior BChE inhibition (IC50 = 13.49 ± 0.44 µM). Kinetic studies revealed mixed-type inhibition for compound 5b (Ki = 10.03 µM, Kis = 36.16 µM), and molecular docking confirmed its stable interactions with AChE’s catalytic and peripheral anionic sites. These findings underscore the potential of cinnamoyl piperidinyl acetate derivatives as promising scaffolds for further optimization and development into effective AD therapeutics.
Similar content being viewed by others
Introduction
Alzheimer’s disease (AD), a progressive neurological disorder, is known for cognitive decline, memory loss, and aberrant behavior. It affects millions of people globally, making it the most common cause of dementia. Amyloid-beta plaque outside the neurons, Neurofibrillary tangles, and neuroinflammation are part of the pathophysiology of AD and lead to the degeneration of cholinergic neurons in the brain1,2,3,4. Targeting the cholinergic system is one of the main therapeutic approaches for controlling the symptoms of AD. According to cholinergic dysfunction, the decline in acetylcholine (ACh), a neurotransmitter essential to brain function, resulted in memory loss and cognitive impairment. ACh is hydrolyzed into acetate by the cholinesterase enzymes named butyrylcholinesterase (BChE) and acetylcholinesterase (AChE)5,6,7.
AChE is mostly found in synaptic clefts and is in charge of controlling ACh levels; however, during the late stages of AD, BChE takes a compensatory function of AChE highlighting the benefits of considering both AChE and BChE as therapeutic targets. FDA-approved AChE inhibitors increase ACh levels including galantamine, rivastigmine, and donepezil. Nevertheless, these inhibitors have limited effectiveness which makes the designing of new molecules necessary7,8.
Carbamate moiety is present in the structure of rivastigmine (Compound A, Fig. 1), a powerful cholinesterase inhibitor to targets AD. Considering the critical role of carbamate moiety, compound B was developed which effectively blocked AChE, and raised the amount of ACh in the A549 cell line, showing encouraging nootropic effects. In the rat brain, compound B efficiently suppresses AChE activity (AChE = 1.266 ng/mL)9. A literature review was performed to identify the optimal scaffold for attaching to the carbamate. In the study by Sheikh et al., chalcone was bioisosterically replaced manually. This work utilized the MolOpt web server to develop new analogs with improved pharmacokinetics and pharmacodynamics. Compound C outperformed the donepezil as a positive control in terms of AChE inhibition with IC50 = 15.3 nM. Additionally, Compound C in vivo assessment against STZ-induced dementia in rats showed that memory improved in the Morris Water Maze test. Compound C was also reported as one of the most potent antioxidant agents (EC50 = 22.83 nM)10. A series of benzylaminochalcone derivatives with different substituents were synthesized and evaluated as inhibitors of AChE. These compounds exhibited IC50 values ranging from 23 to 39 µM (Compound D)11. The N, N-disubstituted carbamate pharmacophore of rivastigmine was combined with an amino chalcone. Compound E displayed a high inhibitory potency against AChE (IC50 = 4.91 µM) and significant antioxidative activity, exceeding that of Trolox by 2.83-fold. Additionally, E inhibited self-induced Aβ1–42 aggregation and Cu2+−induced Aβ1−42 aggregation by 89.5% and 79.7% at 25 µM, respectively. It also acted as a selective monoamine oxidase B inhibitor (IC50 = 0.29 µM) and a selective biometal chelator12.
Regarding the high potency of the mentioned analogs, in the current study, tert-butyl acetate was conjugated with derivatives of cinnamonoyl piperidinyl. After optimizing the molecular length, 17 derivatives with different aromatic ring substituents were synthesized. The inhibitory activities of all derivatives against AChE and BChE were assessed. Furthermore, molecular docking, molecular dynamics simulations, and kinetic studies were performed.

Design of cinnamoyl piperidinyl acetate.
Results and discussion
Chemistry
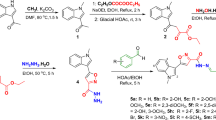
Cinnamic acids were synthesized through a Knoevenagel condensation process involving the reaction of various benzaldehyde derivatives (1a-q; 25 mmol) with malonic acid (compound 2; 30 mmol) in pyridine as the solvent. Following the condensation, β-alanine was incorporated, and the reaction mixture was heated to 100 °C for an appropriate duration to facilitate the formation of the desired product. Upon completion, the solution was allowed to cool to room temperature, after which ice water was added. The pH of the solution was subsequently adjusted to acidic conditions using HCl, promoting the precipitation of the cinnamic acid derivatives (3a-q). The resulting precipitate was filtered, washed with distilled water, and dried in an oven at 60 °C for 12 h.
In the next phase of the synthesis, 1 mmol of the prepared cinnamic acid (compound 3) was dissolved in DMF, and triethylamine was added to the mixture to facilitate coupling. After 15 min, the temperature was lowered to 0 °C, and 1 mmol of TBTU (O-(Benzotriazol-1-yl)-N, N,N’,N’-tetramethyluronium hexafluorophosphate) was gradually introduced into the flask. After 30 min at this temperature, 1 mmol of 3-N-Boc-piperidine4 was added to the reaction mixture and stirred at room temperature. The progress of the reaction was monitored using thin-layer chromatography (TLC), ensuring the formation of the desired intermediate. Following a 24 h reaction period, water was added to the reaction mixture to precipitate the product. The solid products (5a-q) were subsequently filtered, washed with water, and dried in an oven at 60 °C for an additional 12 h.
The structural characterization of the final compounds (5a-q) was extensively verified via 1HNMR and 13CNMR spectroscopy. For instance, compound 5b, a potent AChE inhibitor, exhibited 1HNMR signals at δ 7.18–8.03 ppm, corresponding to the aromatic protons, and the doublet at δ 1.38 ppm, representing the methylene groups. In its 13CNMR spectrum, the amide carbonyl carbon was observed at δ 165 ppm, confirming the successful incorporation of the 2-chloro substitution. Similarly, the 1HNMR spectrum of compound 5q highlighted signals at δ 1.40, 3.82 and 4.04 ppm, indicative of the ethoxy and methoxy functionalities, which align with its superior BChE inhibitory activity. These spectroscopic analyses validate the structures and purities of the synthesized derivatives, supporting their biological activity. Scheme 1 illustrates the stepwise synthesis of substituted cinnamoyl piperidinyl acetate derivatives (5a-q).

Synthesis Procedures of Cinnamoyl Piperidinyl Acetate Derivatives (5a-q).
Biological activities of 5a-q against ache
Table 1 illustrates the potency of 5a-q derivatives against AChE. The unsubstituted analog, 5a, showed an IC50 of 39.42 ± 3.21 µM, confirming that the primary backbone is suitable as an AChE inhibitor. The compound 5b, with a 2-chloro substituent, exhibited the highest inhibitory activity (IC50 = 19.74 µM). This potency can be attributed to the strong electron-withdrawing nature of the chlorine atom and its ortho position, which might enhance interactions with the active site. Similarly, 5c, containing a 2-bromo substituent, displayed significant activity (IC50 = 46.27 µM); however, the larger steric bulk of bromine slightly reduced its potency compared to 5b. Conversely, 5 d, with a 2-methoxy substituent, showed weaker activity (30.58%), suggesting that electron-donating groups at the ortho position disrupt efficient enzyme inhibition.
Derivatives containing substituents at the meta position were synthesized to explore their biological activity. Compound 5e, featuring a 3-fluoro substituent, demonstrated notable potency with an IC50 value of 37.65 µM. The enhanced activity of this compound can be attributed to the electronegativity and small size of fluorine, which produce significant inductive effects that improve the compound’s ability to interact with the target enzyme. In contrast, compound 5f, which possesses a 3-chloro substituent, exhibited reduced potency compared to compound 5b, carrying a 2-chloro substituent. This difference underscores the importance of the positional effect on activity, where the relative placement of substituents significantly influences binding effectiveness. Similarly, compound 5 g, containing a 3-nitro group, showed good activity with an IC50 of 35.13 µM. The strong electron-withdrawing property of the nitro group increases the electron deficiency of the aromatic ring, which might facilitate interactions with active site residues. This effect contrasts with 5 h, which has a 3-methoxy substituent exhibited moderate activity (44.13%). The meta position of the methoxy group appears to have unfavorable effects. Conversely, compound 5 h, featuring a 3-methoxy substituent, displayed moderate activity. The presence of the methoxy group at the meta position appears to have unfavorable effects, suggesting that this substitution may hinder optimal interactions with the enzyme’s active site. In contrast, para-substituted derivatives generally exhibited moderate to low activity compared to their ortho or meta counterparts. For example, 5i and 5j, bearing 4-chloro and 4-bromo groups, showed lower activity (37.09% and 33.91%, respectively) compared to their ortho-substituted counterparts. This trend suggests that para-substituents are less effective at enhancing interactions with critical residues in the active site. Similarly, 5 m and 5n, with para-methoxy and para-ethoxy substituents, exhibited moderate activity (34.85% and 44.41%). The slightly improved hydrophobic interactions of the ethoxy group may explain the better performance of 5n over 5 L.
Di-substituted derivatives provided additional insights into the structure-activity relationships (SAR) of these compounds. Compound 5o, featuring 2,5-dimethoxy groups, exhibited good activity (IC50 = 41.54 µM). In contrast, another derivative, also labeled as 5o, containing 3,4-dimethoxy groups, displayed moderate activity (44.12%), indicating that the positional arrangement of substituents significantly influences potency. Furthermore, compound 5q, which includes a 4-ethoxy-3-methoxy substituent, demonstrated favorable activity (IC50 = 35.23 µM). The observed potency can be attributed to the combination of the ethoxy group’s lipophilicity and the electron-donating properties of the methoxy group. This combination likely enhances favorable interactions within the enzyme’s binding site, optimizing both hydrophobic and electronic contributions to binding affinity.
In summary, both the electronic nature and the position of substituents significantly influence AChE inhibitory activity. Ortho-substituted derivatives, particularly those with electron-withdrawing groups like 2-chloro (5b), exhibited the highest potency, highlighting the importance of favorable electrostatic interactions and positioning for optimal binding. Meta-substituents such as 3-fluoro (5e) and 3-nitro (5 g) also enhanced activity, suggesting that inductive effects at this position support effective enzyme interaction. In contrast, electron-donating groups, especially at the ortho and para positions (e.g., 5 d, 5 h, 5 m), tended to reduce activity, likely due to unfavorable electronic or steric effects. Di-substituted compounds such as 5q (4-ethoxy-3-methoxy) maintained moderate to good activity, indicating that balanced hydrophobic and electronic properties can contribute positively to binding affinity when appropriately positioned.
Biological activities of 5a-q against BChE
The inhibitory activities of the synthesized derivatives against BChE are illustrated in Table 2. The unsubstituted analog, 5a, exhibited an IC50 of 43.69 ± 2.84 µM, suggesting good potency of backbone. Among ortho-substituted derivatives, 5c, with a 2-bromo group, showed the highest potency (IC50 = 14.11 ± 5.78 µM). This result highlights the role of bulk and electronic effects of bromine to enhance potency. Also, 5b, featuring a 2-chlorine group, exhibited strong activity (IC50 = 17.51 ± 4.79 µM). However, 5 d, with a 2-methoxy substituent, displayed slightly reduced activity (IC50 = 44.18 ± 2.16 µM), indicating that the electron-withdrawing effects of substituents at the ortho position positively influence BChE inhibition.
For meta-substituted derivatives, 5e, with a 3-fluoro substituent, exhibited notable activity (IC50 = 15.33 ± 6.53 µM). Fluorine’s small size and electronegativity likely enhance its interaction with the enzyme. In contrast, 5f, containing a 3-chloro substituent, showed poor inhibition (16.67%), confirming that the meta position significantly affects potency. 5 h, with a 3-methoxy group, demonstrated moderate activity (IC50 = 43.76 ± 4.27 µM), similar to 5 d showing the position of methoxy does not affect the activity. Compound 5 g, with a 3-nitro group, exhibited weak activity (33.03%), indicating that the strong electron-withdrawing property of nitro groups at the meta position does not favor binding within the enzyme’s active site.
Among para-substituted derivatives, 5 L, bearing a 4-methyl group, displayed strong inhibitory activity (IC50 = 15.24 ± 2.47 µM). The methyl group’s hydrophobic interactions likely enhance binding affinity. In contrast, 5 m and 5n, featuring 4-methoxy and 4-ethoxy substituents, exhibited moderate activity suggesting that larger substituents at the para position may hinder optimal interactions within the binding pocket. Additionally, 5j, with a 4-bromo group, showed moderate activity (IC50 = 38.10 ± 2.54 µM), confirming that substituents at the para position generally exhibit lower activity compared to their ortho and meta counterparts.
The synthesis of di-substituted derivatives provided additional insights into SAR. Compound 5q, with a 4-ethoxy-3-methoxy combination, demonstrated strong activity (IC50 = 13.49 ± 0.44 µM). This potency is likely due to the synergistic effects of the ethoxy group’s lipophilicity and the methoxy group’s electron-donating properties, which enhance interactions within the active site. Conversely, 5o and 5p exhibited weak activity (35.11% and 33.27%, respectively). The steric bulk of these groups may hinder optimal alignment within the enzyme’s binding site, reducing their potency. Overall, compounds 5c, and 5q emerged as the most potent BChE inhibitor.
Overall, the BChE inhibitory activity of compounds 5a–q revealed that both substitution pattern and electronic properties significantly influenced potency. Ortho-substituted compounds, particularly 5c (2-bromo, IC50 = 14.11 µM) and 5b (2-chloro, IC₅₀ = 17.51 µM), showed the highest activity, suggesting that electron-withdrawing groups at this position enhance binding. Meta-substituents like 3-fluoro (5e) also demonstrated strong activity, while others such as 3-methoxy (5 h) and 3-nitro (5 g) had reduced potency, highlighting the sensitivity of this position. Para-substituted compounds generally exhibited moderate activity, with 5 L (4-methyl, IC50 = 15.24 µM) standing out, likely due to favorable hydrophobic interactions. Di-substituted compound 5q (4-ethoxy-3-methoxy, IC50 = 13.49 µM) emerged as the most potent, indicating that synergistic electronic and lipophilic effects can enhance inhibition. Overall, 5b, 5c, 5e, 5 L, and 5q were the most effective BChE inhibitors, emphasizing the importance of steric and electronic tuning for optimal activity.
Comparison of ache Inhibition vs. BChE Inhibition
The comparison of AChE and BChE inhibitory activities underscores the critical influence of active site characteristics on SAR. AChE’s active site is smaller and more rigid, favoring derivatives with smaller or optimally positioned substituents that enhance electronic interactions. For example, 5b (R: 2-Cl) demonstrated the highest potency against AChE (IC50 = 19.74 µM), attributed to the electron-withdrawing chlorine group in the ortho position, which enhances key interactions with the enzyme’s active site residues. Conversely, bulkier substituents, such as 5c (R: 2-Br), showed reduced potency for AChE (IC50 = 46.27 µM), likely due to steric hindrance within its confined active site.
In contrast, BChE’s larger and more flexible active site accommodates bulkier substituents more effectively, resulting in enhanced inhibitory potency. For instance, 5c (R: 2-Br) exhibited significantly improved inhibition of BChE (IC50 = 14.11 µM) compared to AChE. Similarly, 5q (4-ethoxy-3-methoxy) achieved superior activity against BChE (IC50 = 13.49 µM), benefiting from the bulkier ethoxy group that enhances hydrophobic interactions. These observations highlight a key distinction: while AChE mostly favors smaller substituents for optimal interaction, BChE’s broader active site allows bulkier groups to exploit hydrophobic and steric interactions for improved potency (Fig. 2).

Structural SAR of 5a-p.
Enzyme kinetic studies
Kinetic studies were performed to investigate the inhibitory behavior of compound 5b against AChE. As exhibited in Fig. 3a, compound 5b showed mix-type inhibition. To estimate Ki, the slope of the line was plotted against various inhibitor concentrations, yielding a value of 10.03 µM (Fig. 3b). Moreover, plotting 1/Vmax against different inhibitor concentrations enabled the determination of the secondary inhibition constant (Kis), which was calculated to be 36.16 µM (Fig. 3c).

(a) Line weaver-burk plot of 5b against BChE (b) The secondary plot between the slope of lines and various concentrations of 5b, (c) The secondary plot between 1/Vmax and various concentrations of 5b.
Molecular modeling studies
Computational docking studies were done to assess the interaction of compound 5b with AChE, The reliability of the docking method was validated by redocking AChE with their respective native ligand, achieving RMSD values of less than 2 Å.
AChE binding site comprise different pockets. The peripheral anionic site (PAS), located at the entrance of the active-site gorge, includes key residues such as Tyr124, Trp286, and Tyr295. The catalytic anionic site (CAS) at the bottom of the gorge features the catalytic triad of Ser203, His447, and Glu334. Additionally, the choline-binding site contains residues such as Trp86 and Trp337, further contributing to AChE’s structural and functional properties.
The molecular docking study of 5b with a docking score of −8.419 Kcal/mol is exhibited in Fig. 4, the tert-butyl acetate moiety of compound 5b forms two hydrogen bonds with Gly202 and His447 critical of the enzyme (Fig. 4a and b). Meanwhile, the 2-chlorophenyl group interacts with Phe295 and Arg296 through halogen bonding in the PAS pocket this moiety also showed pi-pi stacking interaction with Trp286. These interactions are consistent with the biological results from the kinetic study, indicating that the compound engages both the CAS and PAS pockets.

Molecular docking analysis of 5b compounds against AChE. (A) 2D interactions of 5b, (B) 3D interactions of 5b.
Molecular dynamic simulations
Molecular dynamics simulations were performed to evaluate the stability of the AChE-5b complex compared to their unbound AChE. The root mean square deviation (RMSD) was monitored to assess structural consistency during the simulations. For apo AChE, RMSD values increased during the initial phase as the protein underwent conformational adjustments. This was followed by a gradual rise to approximately 1.5 Å before stabilizing, with an average RMSD of 1.8 Å. In contrast, the AChE-5b complex exhibited rapid stabilization, with the RMSD plateauing at a lower value of 1.2 Å. This suggests the formation of a stable complex, indicating that compound 5b interacts strongly with AChE and enhances the structural rigidity of the enzyme-inhibitor complex. The low RMSD value supports the potential of 5b as an effective inhibitor (Fig. 5).

RMSD plot of AChE backbone (Apo, blue color) and compound 5b (orange) throughout the 100 ns of the simulation time.
Next, The interactions of compound 5b within the AChE active site were closely monitored during the MD simulation. The tert-butyl acetate moiety formed a stable hydrogen bond with His447, a crucial residue in the catalytic triad at almost all times of the MD run. Additionally, the carbonyl group attached to the piperidine ring formed two hydrogen bonds, mediated by water molecules, with Arg296 and Phe295 (Fig. 6a). Also another H-bound interaction is observed with Tyr124.
The terminal 2-chlorophenyl group is also oriented toward Trp286 Phe338 and Tyr341, which is part of the PAS. To confirm the stability of these interactions throughout the MD simulation and ensure that the complex did not undergo significant rotation within the active site, the timeline representation of interactions was analyzed (Fig. 6b). The interactions were consistently maintained across all snapshots captured during the simulation, further supporting the stability and robustness of the AChE-5b complex.

(A) 2D interaction diagram and (B) timeline interaction of compound 5b -AChE complex, which is responsible for over 30% of MD simulation time.
Notably, the Root Mean Square Fluctuation (RMSF) analysis of compound 5b within the AChE active site further supports this hypothesis. All atoms of 5b recorded RMSF values of less than 2 Å, indicating that the ligand remained firmly anchored within the active site. This stable binding ensured that both critical regions of the enzyme, CAS and PAS, remained occupied throughout the simulation. This reinforces that compound 5b forms a robust interaction with AChE, contributing to its potential as an effective inhibitor (Fig. 7).

RMSF of compound 5b in the AChE active site.
In Silico Drug-Likeness, ADME, and toxicity studies
Compound 5b adheres to Lipinski’s rule of five, showing good drug-like properties with no violations using https://admetlab3.scbdd.com/ (Fig. 8). It demonstrates excellent distribution potential with a high plasma protein binding (PPB) rate of 96.297%. Regarding metabolism, compound 5b is not a substrate for CYP2D6 and does not inhibit CYP1 A2, reducing the risk of metabolism-related adverse effects. Toxicity predictions reveal no AMES toxicity alerts, suggesting that the compound is non-mutagenic (Table 3). These combined in silico findings support the further investigation of compound 5b as a promising lead molecule.

Physicochemical properties of 5b.
Conclusion
This study highlights the potential of cinnamoyl piperidinyl acetates as anti-alzhimer agents. In terms of chemistry, the compounds were efficiently synthesized through a Knoevenagel condensation followed by coupling with substituted piperidines, showing how strategic chemical modifications—such as β-alanine incorporation—can adjust the biological activity. All derivatives were then evaluated as possible AChE and BChE inhibitors, providing valuable insights into the SAR related to their biological activity.
Among the synthesized compounds, 5b, featuring a 2-chloro substitution, exhibited the most potent inhibitory activity against AChE, with an IC50 value of 19.74 µM. This compound also demonstrated significant inhibition of BChE, yielding an IC50 value of 17.51 µM, thereby highlighting its potential as a dual-target agent. The SAR analysis revealed that substitutions at the ortho position with electron-withdrawing groups, such as chlorine, enhanced enzyme inhibition, likely due to improved interactions within the active site through hydrophobic and electronic effects.
Molecular docking and dynamic further highlighted 5b strong and stable binding behavior, with several H- bonding, and π-π stacking interactions stabilizing the complex over time. These findings not only validate the design principles from a chemical view but also provide mechanistic insights valuable to biological applications.
Overall, this study effectively bridges synthetic chemistry with biological activity, offering insights for the development of drug-like candidates aimed at targeting Alzheimer’s disease (AD).
Method and materials
Synthesis of Cinnamic Acid Derivatives (3a-q):
Initially, 25 mmol of benzaldehyde, along with β-alanine, 30 mmol of malonic acid, and pyridine as the solvent, was placed in a round-bottom flask. The mixture was refluxed for 2 h, and the progress of the reaction was monitored using thin-layer chromatography (TLC). Subsequently, the reaction mixture was poured into water, and the pH was adjusted to acidic conditions using hydrochloric acid (HCl). The resulting precipitate was thoroughly washed with water and dried for subsequent use in further steps.
Synthesis of (Boc-1-cinnamoyl piperidin-4-yl)carbamate Derivatives (5a-q):
An amount of 1 mmol of the acid prepared in the previous step was combined with 4 mL of dimethylformamide (DMF) in a suitable container, followed by the addition of triethylamine. The solution was stirred for 20 min and then cooled to 0 °C. At this stage, 1 mmol of the coupling reagent TBTU was gradually added to the mixture. After 30 min, 1 mmol of (Boc-piperidin-4-yl)carbamate was introduced, and the reaction was stirred at room temperature. The progress of the reaction was monitored using thin-layer chromatography (TLC), and the final product was obtained after 24 h.
Chemistry
tert-butyl (1-cinnamoylpiperidin-3-yl) carbamate (5a)
Yield: 77%. Cream powder. 1H NMR (400 MHz, DMSO) δ 7.72 (d, J = 7.2 Hz, 1H), 7.66 (d, J = 7.1 Hz, 1H), 7.47 (d, J = 15.4 Hz, 1H), 7.39 (d, J = 7.5 Hz, 2 H), 7.26 (d, J = 15.4 Hz, 1H), 7.13 (d, J = 15.4 Hz, 1H), 6.96 (dd, J = 11.3, 6.9 Hz, 1H), 4.36–4.26 (m, 1H), 4.09 (d, J = 13.5 Hz, 1H), 3.84–3.74 (m, 1H), 3.73–3.60 (m, 1H), 3.44 (t, J = 10.3 Hz, 1H), 3.29 (s, 1H), 3.05 (d, J = 13.6 Hz, 1H), 1.77 (d, J = 23.3 Hz, 2 H).13C NMR (101 MHz, DMSO) δ 165.01, 155.29, 141.85, 141.69, 135.64, 129.92, 129.19, 128.47, 128.30, 119.02, 118.75, 78.25, 62.48, 49.90, 47.72, 47.24, 47.09, 45.65, 42.49, 30.94, 29.98, 28.70, 25.97, 25.12, 22.90 ppm.
tert-butyl (E)-(1-(3-(2-chlorophenyl)acryloyl)piperidin-3-yl)carbamate (5b)
Dark pink solid; isolated yield: 67%, 1H NMR (400 MHz, DMSO) δ 8.03–7.90 (m, 1H), 7.81–7.76 (m, 1H), 7.51 (d, J = 5.9 Hz, 1H), 7.39 (d, J = 4.3 Hz, 1H), 7.32 (d, J = 15.4 Hz, 1H), 7.18 (d, J = 15.3 Hz, 1H), 6.96 (t, J = 7.7 Hz, 1H), 4.34–4.25 (m, 1H), 4.06 (s, 1H), 3.75 (d, J = 4.0 Hz, 1H), 3.58–3.52 (m, 1H), 3.31 (s, 1H), 3.07 (t, J = 10.4 Hz, 1H), 2.65–2.56 (m, 1H), 1.77 (d, J = 13.6 Hz, 2 H), 1.38 (s, 9 H) ppm.13C NMR (101 MHz, DMSO) δ 164.54, 155.28, 136.76, 133.36, 131.37, 130.26, 128.61, 127.96, 121.99, 78.23, 49.82, 47.95–46.73, 45.69, 42.66, 40.18, 39.97, 39.76, 25.96, 25.04, 22.59, 30.88, 28.65 ppm.
tert-butyl (E)-(1-(3-(2-bromophenyl)acryloyl)piperidin-3-yl)carbamate: (5c)
Light pink solid; isolated yield: 83%, 1H NMR (400 MHz, DMSO) δ 8.01–7.87 (m, 1H), 7.75 (dd, J = 15.2, 2.9 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.41 (t, J = 7.2 Hz, 1H), 7.28 (d, J = 16.4 Hz, 2 H), 6.95 (d, J = 9.5 Hz, 1H), 4.31 (d, J = 12.8 Hz, 1H), 4.07 (d, J = 13.2 Hz, 1H), 3.72 (d, J = 16.0 Hz, 1H), 3.56 (d, J = 6.0 Hz, 1H), 3.30 (s, 1H), 3.06 (t, J = 10.9 Hz, 1H), 2.67–2.53 (m, 1H), 1.77 (d, J = 14.3 Hz, 2 H), 1.27 (s, 9 H) ppm.13C NMR (101 MHz, DMSO) δ 164.48, 155.29, 139.47, 135.07, 133.50, 131.55, 129.10, 128.14, 124.73 122.08 78.22, 40.39, 39.97, 39.77 ppm.
tert-butyl (E)-(1-(3-(2-methoxyphenyl)acryloyl)piperidin-3-yl)carbamate (5 d)
Yield: 80%. Dark Cream powder. 1H NMR (400 MHz, DMSO) δ 7.83–7.63 (m, 3 H), 7.36 (t, J = 7.9 Hz, 1H), 7.14 (d, J = 16.1 Hz, 1H), 7.06 (d, J = 8.3 Hz, 1H), 6.97 (s, 1H), 4.32 (d, J = 10.0 Hz, 1H), 4.04 (d, J = 13.3 Hz, 1H), 3.86 (s, 3 H), 3.73 (d, J = 12.4 Hz, 1H), 3.33 (s, 1H), 3.28–3.18 (m, 1H), 3.02 (d, J = 11.5 Hz, 1H), 2.56 (t, J = 10.9 Hz, 1H), 1.71 (s, 1H), 1.36 (d, J = 31.6 Hz, 9 H) ppm.13C NMR (101 MHz, DMSO) δ 165.28, 157.98, 157.82, 155.62, 155.29, 136.84, 136.42, 131.34, 128.85, 128.31, 123.96, 120.97, 118.81, 112.01, 78.24, 56.02, 55.94, 49.98, 47.79, 47.08, 45.64, 42.50, 30.95, 29.90, 28.65, 25.10, 23.04 ppm.
tert-butyl (E)-(1-(3-(3-fluorophenyl)acryloyl)piperidin-3-yl)carbamate (5e)
Pink solid; isolated yield: 76%, 1H NMR (400 MHz, DMSO) δ 7.65 (dd, J = 37.2, 10.1 Hz, 1H), 7.53–7.42 (m, 3 H), 7.24–7.16 (m, 2 H), 6.95 (d, J = 9.9 Hz, 1H), 4.32 (d, J = 12.5 Hz, 1H), 4.10 (d, J = 13.5 Hz, 1H), 3.80 (d, J = 13.1 Hz, 1H), 3.44 (t, J = 10.4 Hz, 1H), 3.28 (s, 1H), 3.04 (s, 1H), 2.52 (d, J = 1.8 Hz, 1H), 1.79 (d, J = 38.1 Hz, 2 H), 1.36 (d, J = 35.2 Hz, 9 H) ppm. 13C NMR (101 MHz, DMSO) δ 164.54, 155.28, 136.76, 133.36, 131.37, 130.26, 128.61, 127.96, 121.99, 78.23, 49.82, 47.95, 46.73, 45.69, 42.66, 40.18, 39.97, 39.76, 30.88, 29.72, 28.65, 25.96, 25.04, 22.59 ppm.
tert-butyl (E)-(1-(3-(3-chlorophenyl)acryloyl)piperidin-3-yl)carbamate (5f):
Yield: 89%. Cream powder. 1H NMR (400 MHz, DMSO) δ 7.88 (d, J = 38.1 Hz, 1H), 7.62 (d, J = 17.4 Hz, 1H), 7.42 (d, J = 5.1 Hz, 3 H), 7.29 (dd, J = 50.9, 15.3 Hz, 1H), 6.99–6.93 (m, 1H), 4.31 (d, J = 12.5 Hz, 1H), 4.11 (d, J = 13.9 Hz, 1H), 3.74 (dd, J = 47.8, 12.1 Hz, 1H), 3.51–3.39 (m, 1H), 3.29 (s, 1H), 3.04 (t, J = 11.6 Hz, 1H), 2.57 (t, J = 11.1 Hz, 1H), 1.79 (d, J = 38.0 Hz, 2 H), 1.32 (s, 9 H) ppm.13C NMR (101 MHz, DMSO) δ 134.10, 130.98, 129.52, 127.53, 120.69, 78.28, 48.33, 46.74, 42.57, 40.59, 40.39, 40.18, 39.97, 39.76, 39.55, 39.34, 30.91, 28.69 ppm.
tert-butyl (E)-(1-(3-(3-nitrophenyl)acryloyl)piperidin-3-yl)carbamate (5 g)
Beige solid; isolated yield: 87%, 1H NMR (400 MHz, DMSO) δ 8.64–8.56 (m, 1H), 8.20 (dt, J = 7.2, 1.3 Hz, 1H), 7.69 (t, J = 8.0 Hz, 1H), 7.59 (d, J = 15.1 Hz, 1H), 7.51 (d, J = 15.5 Hz, 1H), 7.38 (d, J = 15.5 Hz, 1H), 7.00–6.94 (m, 1H), 4.32 (d, J = 12.5 Hz, 1H), 4.14 (d, J = 13.5 Hz, 1H), 3.84 (d, J = 13.2 Hz, 1H), 3.69 (s, 1H), 3.29 (s, 1H), 3.13–2.99 (m, 1H), 2.58 (t, J = 11.1 Hz, 1H), 1.75 (s, 2 H), 1.30 (s, 9 H) ppm. 13C NMR (101 MHz, DMSO) δ 164.57, 148.79, 139.37, 137.59, 134.90, 130.68, 124.18, 123.16, 122.13, 78.28, 47.77, 47.17, 45.67, 40.58, 40.37, 40.16, 39.95, 39.74, 39.54, 39.33, 30.89, 28.67, 25.11 ppm.
tert-butyl (E)-(1-(3-(3-methoxyphenyl)acryloyl)piperidin-3-yl)carbamate (5 h)
Cream solid; isolated yield: 90%, 1H NMR (400 MHz, DMSO) δ 7.42 (s, 1H), 7.32 (d, J = 7.5 Hz, 1H), 7.28 (d, J = 4.5 Hz, 1H), 7.26–7.10 (m, 3 H), 6.93 (d, J = 2.4 Hz, 1H), 4.39–4.26 (m, 1H), 4.10 (d, J = 13.6 Hz, 1H), 3.85 (s, 1H), 3.80 (s, 3 H), 3.73 (s, 1H), 3.22 (s, 1H), 3.04 (s, 1H), 2.56 (t, J = 11.0 Hz, 1H), 1.79 (d, J = 37.1 Hz, 2 H), 1.40 (s, 9 H) ppm.13C NMR (101 MHz, DMSO) δ 141.77 (d, J = 22.9 Hz), 130.21, 121.09 (d, J = 18.8 Hz), 115.87, 113.12 (d, J = 20.3 Hz), 78.26, 55.68, 48.08–46.47 (m), 40.49 (d, J = 21.0 Hz), 40.17, 39.97, 39.76, 39.55, 39.34, 28.69 ppm.
tert-butyl (E)-(1-(3-(4-chlorophenyl)acryloyl)piperidin-3-yl)carbamate (5i)
Yield: 81%. Cream powder. 1H NMR (400 MHz, DMSO) δ 7.77 (d, J = 8.2 Hz, 1H), 7.70 (d, J = 8.2 Hz, 1H), 7.47 (s, 1H), 7.44 (d, J = 4.5 Hz, 1H), 7.29 (d, J = 15.4 Hz, 1H), 7.16 (d, J = 15.4 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 4.32 (d, J = 10.7 Hz, 1H), 4.09 (d, J = 13.5 Hz, 1H), 3.82–3.74 (m, 1H), 3.68 (d, J = 12.8 Hz, 1H), 3.44 (d, J = 21.2 Hz, 1H), 3.28 (s, 1H), 3.04 (t, J = 11.5 Hz, 1H), 1.85–1.70 (m, 2 H), 1.40 (s, 9 H) ppm.13C NMR (101 MHz, DMSO) δ 164.86, 155.59, 155.28, 140.45, 140.29, 134.64, 134.32, 130.22, 130.05, 129.19, 119.91, 119.65, 78.24, 49.87, 47.69, 47.24, 47.07, 45.64, 42.51, 30.92, 30.00, 28.69, 25.97, 25.11, 22.86 ppm.
tert-butyl (E)-(1-(3-(4-bromophenyl)acryloyl)piperidin-3-yl)carbamate (5j):
Yield: 77%. cream powder. 1H NMR (400 MHz, DMSO) δ 7.70 (d, J = 8.2 Hz, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.59 (d, J = 8.0 Hz, 2 H), 7.44 (d, J = 15.3 Hz, 1H), 7.30 (d, J = 15.4 Hz, 1H), 7.18 (d, J = 15.4 Hz, 1H), 6.94 (d, J = 8.3 Hz, 1H), 4.40–4.22 (m, 1H), 4.13–3.74 (m, 1H), 3.72–3.38 (m, 1H), 3.30 (d, J = 12.8 Hz, 1H), 2.80 (dt, J = 186.8, 11.1 Hz, 2 H), 1.89–1.68 (m, 2 H), 1.35 (d, J = 36.2 Hz, 9 H) ppm. 13C NMR (101 MHz, DMSO) δ 164.86, 155.60, 155.28, 140.54, 140.38, 134.98, 132.12, 130.47, 130.30, 123.10, 119.99, 119.72, 78.25, 49.87, 47.69, 47.24, 47.07, 45.65, 42.52, 30.91, 30.00, 28.70, 25.10, 22.85 ppm.
tert-butyl (E)-(1-(3-(4-nitrophenyl)acryloyl)piperidin-3-yl)carbamate (5k)
Dark beige solid; isolated yield: 75%, 1H NMR (400 MHz, DMSO) δ 8.23 (d, J = 8.3 Hz, 1H), 8.01 (d, 1H), 7.93 (d, 1H), 7.55 (d, J = 15.2 Hz, 1H), 7.48 (d, J = 15.6 Hz, 1H), 7.36 (d, J = 15.5 Hz, 1H), 6.95 (d, J = 8.6 Hz, 1H), 4.36–4.23 (m, 1H), 4.13–4.00 (m, 1H), 3.80 (d, J = 13.3 Hz, 1H), 3.73–3.62 (m, 1H), 3.32 (m, 1H), 3.09 (d, J = 11.5 Hz, 1H), 2.69–2.53 (m, 1H), 1.75 (d, 2 H), 1.28 (d, 9 H) ppm. 13C NMR (101 MHz, DMSO) δ 164.44, 155.47, 147.89, 142.29, 139.21, 129.43, 124.30, 123.54, 78.29, 49.92, 47.15, 45.74, 42.60, 40.11, 39.90, 39.48, 30.84, 29.92, 28.66, 25.04, 22.79 ppm.
tert-butyl (E)-(1-(3-(p-tolyl)acryloyl)piperidin-3-yl)carbamate (5 L)
Beige solid; isolated yield: 71%, 1H NMR (400 MHz, DMSO) δ 7.58 (dd, J = 26.9, 7.8 Hz, 2 H), 7.44 (d, J = 15.4 Hz, 1H), 7.21 (d, J = 7.5 Hz, 2 H), 7.07 (d, J = 15.4 Hz, 1H), 6.98–6.92 (m, 1H), 4.43–4.21 (m, 1H), 4.08 (d, J = 13.5 Hz, 1H), 3.79 (d, J = 12.2 Hz, 1H), 3.69 (s, 1H), 3.28 (s, 1H), 3.03 (s, 1H), 2.57 (d, J = 11.6 Hz, 1H), 2.32 (s, 3 H), 1.78 (d, J = 38.5 Hz, 2 H), 1.36 (d, J = 29.1 Hz, 9 H) ppm. 13C NMR (101 MHz, DMSO) δ 141.82 (d, J = 15.8 Hz), 129.79, 128.38 (d, J = 17.4 Hz), 78.24, 48.27–46.59 (m), 40.59, 40.27 (d, J = 21.0 Hz), 39.96, 39.75, 39.54, 39.33, 28.70, 21.42 ppm.
tert-butyl (E)-(1-(3-(4-methoxyphenyl)acryloyl)piperidin-3-yl)carbamate (5 m)
Cream solid; isolated yield: 93%, 1H NMR (400 MHz, DMSO) δ 7.68 (s, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.44 (d, J = 15.3 Hz, 1H), 7.10 (d, J = 15.4 Hz, 1H), 6.94 (s, 3 H), 4.38–4.28 (m, 1H), 4.08 (d, J = 13.5 Hz, 1H), 3.79 (s, 3 H), 3.73 (s, 1H), 3.46–3.36 (m, 1H), 3.27 (s, 1H), 3.02 (s, 1H), 2.56 (d, J = 11.3 Hz, 1H), 1.83 (s, 2 H), 1.40 (s, 9 H).13C NMR (101 MHz, DMSO) δ 165.24, 160.80, 155.29, 141.67, 130.02, 128.25, 116.14, 114.61, 78.23, 55.71, 47.41, 45.58, 40.28, 39.96, 39.76, 30.98, 30.08, 28.70, 25.16, 22.97 ppm.
tert-butyl (E)-(1-(3-(4-ethoxyphenyl)acryloyl)piperidin-3-yl)carbamate (5n)
Cream solid; isolated yield: 67%, 1H NMR (400 MHz, DMSO) δ 7.37–7.31 (m, 3 H), 6.98–6.91 (m, 4 H), 4.23 (q, J = 6.5 Hz, 1H), 4.06 (q, J = 7.0 Hz, 3 H), 3.68 (d, J = 86.8 Hz, 1H), 3.35 (s, 2 H), 2.97 (d, J = 11.2 Hz, 1H), 2.69 (d, J = 12.5 Hz, 1H), 1.84 (dt, J = 9.6, 5.2 Hz, 2 H), 1.38–1.33 (m, 12 H) ppm. 13CNMR (101 MHz, DMSO) δ 169.57, 159.82, 155.33, 129.42, 128.40, 114.38, 78.25, 63.62, 47.17, 40.59, 40.38, 40.17, 39.96, 39.76, 39.55, 39.34, 28.69, 15.03 ppm.
tert-butyl (E)-(1-(3-(2,5-dimethoxyphenyl)acryloyl)piperidin-3-yl)carbamate (5o)
Cream solid; isolated yield: 92%, 1H NMR (400 MHz, DMSO) δ 7.75–7.69 (m, 1H), 7.23 (d, J = 6.4 Hz, 1H), 7.16 (d, J = 15.9 Hz, 1H), 7.01 (s, 1H), 6.95 (s, 1H), 6.93 (d, J = 2.9 Hz, 1H), 4.32 (d, J = 11.0 Hz, 1H), 4.08 (d, J = 13.7 Hz, 1H), 3.87 (d, J = 11.2 Hz, 1H), 3.80 (s, 3 H), 3.75 (s, 3 H), 3.28 (s, 1H), 3.16 (s, 1H), 3.03 (s, 1H), 2.57 (d, J = 11.3 Hz, 1H), 1.75 (s, 2 H), 1.37 (d, 9 H) ppm.13C NMR (101 MHz, DMSO) δ 165.23, 155.62, 153.60, 152.32, 136.39, 124.58, 119.05, 116.82, 113.23, 112.91, 78.25, 56.49–55.57, 47.16, 40.58, 40.27, 39.96, 39.75, 39.54, 39.33, 28.66 ppm.
tert-butyl (E)-(1-(3-(3,4-dimethoxyphenyl)acryloyl)piperidin-3-yl)carbamate (5p)
White solid; isolated yield: 87%, 1H NMR (400 MHz, DMSO) δ 7.41 (s, 1H), 7.33 (d, J = 26.2 Hz, 2 H), 7.16 (d, J = 20.1 Hz, 2 H), 6.95 (s, 1H), 4.34 (d, J = 12.1 Hz, 1H), 4.12 (d, J = 13.2 Hz, 1H), 3.89 (d, J = 11.1 Hz, 1H), 3.82 (s, 3 H), 3.78 (s, 3 H), 3.75 (s, 1H), 3.31 (s, 1H), 3.03 (s, 1H), 2.55 (d, J = 11.1 Hz, 1H), 1.75 (s, 2 H), 1.34 (d, 9 H) ppm.13C NMR (101 MHz, DMSO) δ 165.26, 149.32, 142.17, 128.44, 122.80, 116.12, 111.90, 110.55, 78.25, 55.96, 50.16, 47.99, 47.23, 45.53, 40.16, 39.95, 39.74, 39.53, 30.98, 30.10, 28.68, 25.21, 23.28 ppm.
tert-butyl (E)-(1-(3-(4-ethoxy-3-methoxyphenyl)acryloyl)piperidin-3-yl)carbamate (5q)
Cream solid; isolated yield: 67%, 1H NMR (400 MHz, DMSO) δ 7.42 (d, J = 15.2 Hz, 1H), 7.33 (d, J = 25.6 Hz, 1H), 7.20–6.99 (m, 3 H), 6.93 (s, 1H), 4.41–4.24 (m, 1H), 4.12 (d, J = 13.7 Hz, 1H), 4.04 (q, J = 7.0 Hz, 2 H), 3.89 (d, J = 11.5 Hz, 1H), 3.82 (s, 3 H), 3.79 (s, 1H), 3.30 (d, J = 11.6 Hz, 1H), 3.08 (d, J = 37.0 Hz, 1H), 2.51 (d, J = 1.9 Hz, 1H), 1.85–1.69 (m, 2 H), 1.40 (t, 3 H), 1.35–1.31 (m, 9 H) ppm. 13C NMR (101 MHz, DMSO) δ 165.27, 149.88, 149.39, 142.19, 128.34, 122.79, 112.74, 78.24, 64.11, 56.06, 50.15, 47.99, 47.10, 45.52, 42.56, 40.38, 40.17, 39.96, 39.75, 30.99, 30.55, 29.70, 28.68, 25.23, 23.28, 15.13 ppm.
Screening of ache and BChE inhibitory activity
Cholinesterase inhibitory activities of all derivatives were assessed using the modified Ellman method using butyrylcholinesterase (BChE, E.C. 3.1.1.8, from horse serum), and acetylcholinesterase (AChE, E.C. 3.1.1.7, Type V-S, lyophilized powder, from electric eel, 1000 units)6,13,14.
Enzyme kinetic studies
The mode of inhibition of the most active compound 5b was investigated against AChE in different concentrations of acetylthiocholine substrate (0.1–1 mM) as substrate. A Lineweaver–Burk plot was generated to identify the type of inhibition and the Michaelis–Menten constant (Km) value was determined from the plot between the reciprocal of the substrate concentration (1/[S]) and reciprocal of enzyme rate (1/V) over the various inhibitor concentrations15.
Molecular Docking
The molecular docking of the 5b was conducted using the Schrödinger Suites Maestro molecular modeling platform36. X-ray crystallographic structures of AChE were obtained from the RCSB Protein Data Bank (www.rcsb.org), with a PDB ID of 4EY716,17.
MD simulations
The starting model was obtained by imposing the induced fit docking to AChE (4EY7). MD simulations were conducted using Desmond v5.3 of Schrodinger’s suit maestro according to previously reported procedures18,19.
Data availability
The datasets generated and/or analyzed during the current study are available in the Worldwide ProteinData Bank with the PDB ID of the 4EY7 repository.
References
Breijyeh, Z. & Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules ;25(24). (2020).
Yazdani, M. et al. 5,6-Diphenyl triazine-thio Methyl Triazole hybrid as a new Alzheimer’s disease modifying agents. Mol. Diversity. 24 (3), 641–654 (2020).
Edraki, N. et al. N-(2-(Piperazin-1-yl)phenyl)arylamide derivatives as β-Secretase (BACE1) inhibitors: simple synthesis by Ugi Four-Component reaction and biological evaluation. Arch. Pharm. 348 (5), 330–337 (2015).
Haghighijoo, Z. et al. N-Cyclohexylimidazo[1,2-a]pyridine derivatives as multi-target-directed ligands for treatment of Alzheimer’s disease. Bioorg. Chem. 103, 104146 (2020).
Oliyaei, N., Moosavi-Nasab, M., Tanideh, N. & Iraji, A. Multiple roles of Fucoxanthin and Astaxanthin against Alzheimer’s disease: their Pharmacological potential and therapeutic insights. Brain Res. Bull. 193, 11–21 (2023).
Saeedi, M. et al. Synthesis and bio-evaluation of new multifunctional methylindolinone-1,2,3-triazole hybrids as anti-Alzheimer’s agents. J. Mol. Struct. 1229, 129828 (2021).
Sadeghian, B. et al. Design, synthesis and biological activity evaluation of novel carbazole-benzylpiperidine hybrids as potential anti alzheimer agents. J. Mol. Struct. 1221, 128793 (2020).
Iraji, A., Khoshneviszadeh, M., Firuzi, O., Khoshneviszadeh, M. & Edraki, N. Novel small molecule therapeutic agents for alzheimer disease: focusing on BACE1 and multi-target directed ligands. Bioorg. Chem. 97, 103649 (2020).
Agha, K. A. et al. Novel Sunifiram-carbamate hybrids as potential dual acetylcholinesterase inhibitor and NMDAR co-agonist: simulation-guided analogue design and Pharmacological screening. J. Enzyme Inhib. Med. Chem. 37 (1), 1241–1256 (2022).
Sharma, P. et al. Recent development of novel Aminoethyl-Substituted Chalcones as potential drug candidates for the treatment of Alzheimer’s disease. Molecules 28 (18), 6579 (2023).
Tran, T-D. et al. Synthesis of novel Chalcones as acetylcholinesterase inhibitors. Appl. Sci. 6 (7), 198 (2016).
Xiao, G. et al. Design, synthesis and biological evaluation of 4′-aminochalcone-rivastigmine hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 25 (3), 1030–1041 (2017).
Karimi Askarani, H. et al. Design and synthesis of multi-target directed 1,2,3-triazole-dimethylaminoacryloyl-chromenone derivatives with potential use in Alzheimer’s disease. BMC Chem. 14 (1), 64 (2020).
Saeedi, M. et al. Design and synthesis of selective acetylcholinesterase inhibitors: Arylisoxazole-Phenylpiperazine derivatives. Chem. Biodivers. 16 (2), e1800433 (2019).
Pourtaher, H., Mohammadi, Y., Hasaninejad, A. & Iraji, A. Highly efficient, catalyst-free, one-pot sequential four-component synthesis of novel spiroindolinone-pyrazole scaffolds as anti-Alzheimer agents: in Silico study and biological screening. RSC Med. Chem. 15 (1), 207–222 (2024).
Noori, M. et al. Phenyl-quinoline derivatives as lead structure of cholinesterase inhibitors with potency to reduce the GSK-3β level targeting Alzheimer’s disease. Int. J. Biol. Macromol. 253 (Pt 7), 127392 (2023).
Pourtaher, H., Hasaninejad, A., Zare, S., Tanideh, N. & Iraji, A. The anti-Alzheimer potential of novel spiroindolin-1,2-diazepine derivatives as targeted cholinesterase inhibitors with modified substituents. Sci. Rep. 13 (1), 11952 (2023).
Pourtaher, H., Hasaninejad, A. & Iraji, A. Design, synthesis, in Silico and biological evaluations of novel polysubstituted pyrroles as selective acetylcholinesterase inhibitors against Alzheimer’s disease. Sci. Rep. 12 (1), 15236 (2022).
Nazarian, A. et al. Anticholinesterase activities of novel isoindolin-1,3-dione-based acetohydrazide derivatives: design, synthesis, biological evaluation, molecular dynamic study. BMC Chem. 18 (1), 64 (2024).
Funding
The authors wish to thank the support of the Vice-Chancellor for Research of Shiraz University of Medical Sciences (grant number = IR.SUMS.REC.1404.046).
Author information
Authors and Affiliations
Contributions
M.E. synthesized compounds and contributed to the characterization of compounds. A.I performed in silico and biological studies. S.H and M.M supervised the synthetic part of the study. All authors read and approved the final version of the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Esmkhani, M., Javanshir, S., Iraji, A. et al. Design and evaluation of substituted cinnamoyl piperidinyl acetate derivatives as potent cholinesterase inhibitors. Sci Rep 15, 19346 (2025). https://doi.org/10.1038/s41598-025-04266-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-04266-z
