
Abstract
Chromatophores provide an excellent model to study organelle transport as they specialize in the translocation of pigment granules in response to defined environmental signals. Our previous study revealed low-intensity light-induced xanthosomes aggregation is directly mediated by Opsin 3 in xanthophores of large yellow croaker (Larimichthys crocea). Herein, we report an incidental observation of prolonged (more than 40 min) high-intensity white light (HIWL) (10,000 lux, 3.86 mW/cm2) exposure induce xanthosomes dispersion. After dispersion by HIWL exposure, the xanthosomes remains the ability to aggregate under low-intensity light conditions. Both light-emitting diode (LED) sources at different wavelengths (blue light, λmax = 480 nm; red light, λmax = 686 nm) with identical brightness (10,000 lux) but varying absolute irradiance levels (3.86–22.2 mW/cm2) can induce xanthosome dispersion. Ex vivo illumination and pharmacological experiments on xanthophores revealed that HIWL-induced xanthosomes dispersion is irrelevant to signaling pathways typically associated with xanthosome movement, but mediated by microtubule depolymerization, which is due to extraordinary high level of intracellular Ca2+ released from IP3R calcium channel in endoplasmic reticulum. Interestingly, such a mechanism was also presented in HeLa and HEK293T cells exposed to HIWL. In summary, our results expand our understanding of the impact of high-intensity light on intracellular organelle transport and cytoskeleton.
Similar content being viewed by others
Introduction
The extraordinarily dynamic nature of intracellular organelles is critical in establishing and maintaining intracellular organization and cellular function. Motility is regulated by the activity of organelle-associated motor proteins, kinesins, dyneins and myosins, which move cargo along polar microtubule (MT) and microfilament (MF)1. Over recent years, given the visible nature of pigment granules, chromophores have become an excellent model for understanding the molecular mechanisms of organelle dynamics2.
Among the various types of chromatophores, melanophores—which contain melanin-filled organelles called melanosomes—are the most extensively studied3,4,5,6,7. The transport and translocation of melanosomes within melanophores are orchestrated by elements of the cytoskeleton, particularly MT and MF, along with their associated molecular motors3,4,5,6,7. During pigment dispersal, the molecular motor kinesin facilitates the outward transport of melanin granules along MT towards the periphery of the cell4, after which melanosomes are further translocated along randomly oriented MF5,7. Conversely, during pigment aggregation, the motor protein dynein is responsible for hauling melanosomes back toward the perinuclear region via MT6. The xanthophores are dendritic chromatophores with a yellow tint8. Under electron microscopy, Fujii et al. observed pigment granules within the xanthophores of the goby (Chusmichthys) as globular structures, with diameters comparable to those of melanosomes8. MT were proposed to facilitate the formation and maintenance of the final state of pigment aggregation within xanthophores, suggesting a structural role similar to that observed in melanophores9,10. This finding highlights the functional similarity between xanthophores and melanophores in the intracellular movement of pigment granules, underscoring a conserved mechanism of pigment translocation across chromatophore types.
The large yellow croaker (Larimichthys crocea), as indicated by its common name, exhibits distinctive golden-yellow coloration particularly in the ventral skin11. Our previous research demonstrated that the dispersion of pigment granules, or xanthosomes, within xanthophores is primarily responsible for the yellow coloration of the croaker’s skin12,13,14. Notably, xanthosomes disperse completely in darkness but rapidly aggregate under low-intensity white light (LIWL, 150 lux, 0.04 mW/cm2), with the blue light component (480 nm) being primarily responsible for this effect. This photoreactivity suggests that xanthophores possess intrinsic photosensitivity, enabling dynamic responses to ambient light conditions. We have demonstrated that the dispersion of xanthosomes in large yellow croaker is mediated by the MSH (Melanocyte-stimulating hormone)-induced cAMP-PKA signaling pathway, with elevated cAMP levels leading to xanthosome dispersion12. Conversely, the aggregation of xanthosomes is governed by the Opsin 3-mediated Ca2⁺-PKC signaling pathway15. In addition, the movement of xanthosomes is highly dependent on the cytoskeletal filaments, including microfilaments and microtubules15. These results suggest that xanthophore of large yellow croaker can be used to study the effects of light on transportation of intracellular organelles.
The negative effects of high-intensity and excessive light exposure on cells, such as cell apoptosis induced by endoplasmic reticulum (ER) stress, have increasingly drawn attention16,17. Cell apoptosis is regulated by Ca2+ signaling at the ER, with IP3R mediating Ca2+ release from the ER to the cytoplasm and mitochondria, crucial for transducing death signals18,19. However, little is known about the effect of high-intensity and excessive light exposure on transportation of intracellular organelles. Here we report an incidental observation of prolonged high-intensity white light (HIWL) (10,000 lux, 3.86 mW/cm2) exposure (more than 40 min) induced xanthosomes dispersion in large yellow croaker. HIWL exposure has activated IP3 receptors (IP3R) on ER, leading to an acute increase in intracellular Ca2⁺ concentration, which has subsequently induced microtubule depolymerization and xanthosomes dispersion. We further explore whether this mechanism extends to human cell lines. Our findings may offer a scientific basis for the potential application of high-intensity light in suppressing tumor cell proliferation and progression.
Materials and methods
Experimental fish
Adult large yellow croaker (body length: 36.2 ± 0.7 cm; body weight 716.7 ± 11.9 g) were purchased from a seafood market in Xiamen, Fujian province, China. Fish were temporarily maintained in indoor plastic tanks with fresh seawater at the salinity of 30, a temperature of 25 °C, and a photoperiod of 14L:10D for at least 24 h to acclimate to the experimental environment. Before sampling, the fish were anaesthetized with 0.01% MS222 (Sigma-Aldrich, USA). All experiment protocols were approved by the Institutional Animal Care and Use Committee of Xiamen University; IACUC: XMULAC20230011.
Light source parameters
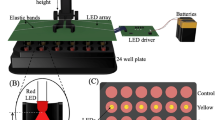
The light sources used in both in vivo and various cells light exposure experiments were all light-emitting diodes (LEDs). The absolute irradiances were as follows: LIWL (150 lux) at 0.04 mW/cm2; HIWL (5000 lux) at 1.93 mW/cm2, (8,000 lux) at 3.09 mW/cm2, (10,000 lux) at 3.86 mW/cm2 and (30,000 lux) at 11.59 mW/cm2; high-intensity blue light (10,000 lux) at 7.11 mW/cm2; high-intensity red light (10,000 lux) at 22.20 mW/cm2. The two types of white light were provided by different light sources. The spectrogram of LIWL, HIWL, blue light (λmax = 480 nm) and red light (λmax = 686 nm) were measured using a fiber optic spectrometer (USB2000 + , Ocean Optics, USA) (Supplemental Fig. S1). The absolute irradiance of HIWL, high-intensity blue light and high-intensity red light were measured using a power and energy meter (PM400, Thorlabs, USA) with a power sensor (S120VC, Thorlabs, USA), while the absolute irradiance of LIWL was measured using a micro-power meter (XM0120, Xunmiao, China). The light intensity and absolute irradiance described in the text were measured at the fish skin and various cells, with values obtained by controlling the distance between the light source and the respective targets.
In vivo illumination experiments
The adult large yellow croakers were first maintained in the dark for 1 h to obtain the experimental fishes with golden yellow ventral skin. Then golden yellow fishes were anaesthetised with 0.01% MS222 (Sigma-Aldrich, USA) and their ventral skins were exposed using light sources of different light intensities (150, 0.04 mW/cm2; 5,000, 1.93 mW/cm2; 8,000, 3.09 mW/cm2; 10,000, 3.86 mW/cm2; 30,000 lux, 11.59 mW/cm2). The images of ventral skins were quickly taken using a camera (SONY, DSC-HX300, Japan). Seven fish were used for each treatment.
Collection and culture of xanthophore
Scales with xanthophores were isolated from the ventral skin and rinsed in 1 × PBS (Gibco, USA) three times. Then, scales were rinsed twice with Dulbecco’s modified eagle medium (DMEM) (Gibco, USA). Under the microscope, the scales, which were covered with abundant xanthophores, were selected and moved to a 24-well cell culture plate (NEST, USA). The xanthophores were maintained in DMEM (Gibco, USA)20,21 supplemented with 1% penicillin–Streptomycin-Gentamicin (Solarbio, China) at 28 °C with 5% CO2.
Ex vivo illumination experiments
Scales with xanthophores were isolated from the ventral skin and transferred to phenol red-free 1 × PBS (Gibco, USA) to prevent potential interference of phenol red with monochromatic light treatments. The light exposure experiments were conducted under dark conditions using LED monochromatic light sources. The images were acquired using an inverted microscope (Leica DMi1, Germany). Throughout the illumination experiments, xanthophores were maintained at 28 °C and 5% CO2. Each treatment was repeated 3 times, and in each replicate, at least 20 xanthophores on scales collected from 2 fish were analysis.
Pharmacological strategies
To decipher the key of HIWL illumination-induced xanthosomes dispersion, morphology assay of xanthosomes after drug application blocking or triggering step by step transduction events were done. Investigations were done to decipher the implication of (i) PKC and Ca2+; (ii) PKA and cAMP; (iii) cytoskeleton correlation in the HIWL illumination-induced xanthosomes dispersion in large yellow croaker. The following drugs were employed: Phorbol-12-myristate-13-acetate (PMA), PKC activator22,23,24; H 89 2HCl, PKA inhibitor25,26; BAPTA-AM, calcium chelating agent27,28,29; Paclitaxel, microtubule-stabilizing agent30,31; Nocodazole, microtubule depolymerizer; A23187, Ca2+ ionophore32; 2-Aminoethyl diphenylborinate (2-APB), cell-permeable inhibitor of IP3R33,34,35. DMSO/EtOH were used as control as the drugs were diluted in these solvents. Illumination conditions were selected according to different experimental requirements: xanthosomes were aggregated in LIWL and dispersed in dark. The light conditions and time during treatment were selected according to different drugs, and after the treatment, xanthophores were transferred to different light conditions and images were collected by inverted microscope (Leica, DMi1, Germany). During drug treatment, xanthophores were maintained at 28 °C and 5% CO2. Each treatment was repeated 3 times, and in each replicate, at least 20 xanthophores on scales collected from 2 fish were analysis. Drugs information and concentrations are summarized in Supplemental Table S1.
Cell culture
Cell culture materials were purchased from Life Technologies (USA) unless specified. HeLa cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), penicillin and streptomycin at 37 °C with 5% CO2.
Intracellular Ca2+ detection
Fluo4-AM is a membrane-permeant compound capable of indicating intracellular Ca2+. HeLa cells were exposed to LIWL or HIWL for 1 h, followed by staining with Fluo4-AM (Beyotime, China). In the IP3R calcium channel blocking experiment, HeLa cells were pre-treated with 100 μM 2-APB (MCE, USA) for 30 min, DMSO was used as the control. The labeled cells were subsequently detected by fluorescence microscope (DM 2500, Leica, Germany). Quantification of Ca2+ fluorescence signals (Fluo4) was measured using ImageJ software (NIH, Bethesda, Maryland, USA)36.
Confocal microscopy
HeLa cells seeded on glass coverslips were illuminated by LIWL and HIWL, fixed with acetone (pre-cooling) at room temperature for 5 min and incubated with blocking solution (5% BSA) for 1 h, followed by 1 h incubation with primary antibodies (Anti-alpha Tubulin, abcam, ab7291, USA) (1:250) at 37 °C. For visualization, fluorescently labeled secondary antibodies (Alexa Fluor® 488, abcam, ab150113, USA) (1:800) were used for 1 h at 37 °C. Cells were further incubated with a nuclear dye DAPI (Beyotime, China) for 5 min. Confocal images were acquired using a confocal microscope (Zeiss, LSM780, Germany).
Statistical analysis
The criteria for determining xanthosome distribution within xanthophores were as follows:
Aggregated state: Defined by spherical xanthosomes clustered near the cell center.
Dispersed state: Defined by xanthosomes distributed diffusely throughout the cytoplasm.
The percentage of aggregated xanthosomes (A1) was calculated as:
The percentage of dispersed xanthosomes (A2) was calculated as:
where A aggregation and A dispersion denote the number of xanthophore with xanthosomes aggregation and dispersion, respectively, and A totality denote the total number of xanthophore. All the experimental data were obtained from at least three independent experiments.
The data were presented as mean ± SEM. The analysis of variance to compare the means of two or more than two groups was performed by t-tests/one‐way/two-way ANOVA analysis. Differences were considered significant when the P value < 0.05. All analyses were carried out with GraphPad Prism Version 8.0 software (GraphPad Software, USA).
Results
High-intensity white light induced xanthosomes dispersion in ventral skin of L. crocea
In the absence of light, the ventral skin of the croaker exhibited a yellow coloration. Following 5 min of exposure to LIWL, the ventral skin color shifted to white and remained so under continued illumination (Fig. 1A). However, when exposed to HIWL, the ventral skin gradually changed back to yellow after approximately 40 min (Fig. 1B). Notably, the skin beneath the pelvic fins remained white, suggesting that there is a threshold of light intensity required to induce the color change from white to yellow (Fig. 1B, C). As expected, the color change from white to yellow was due to the dispersion of xanthosomes (Fig. 1D). To determine whether xanthophores remained viable after prolonged exposure to HIWL, the fish were re-exposed to LIWL after 40-min exposure to HIWL. Under LIWL, the ventral skin color changed from yellow back to white within 4 h (Fig. 1E), indicating that xanthosomes dispersion is reversible. The movement of xanthosomes was in correspondence with the color change (Fig. 1F). To determine the light intensity threshold for inducing xanthosome dispersion, we examined the response under varying illumination conditions. The results showed that when the illuminance reached 10,000 lux and the irradiance reached 3.86 mW/cm2, the proportion of dispersed xanthosomes increased significantly (Fig. 2). Although low-heat LED light sources were used throughout the experiment, temperature remains a significant variable that needs to be taken into account. To address this, we continuously monitored the temperature using a thermal imaging camera during the entire in vivo light exposure experiment and did not observe any significant temperature increase (Supplemental Fig. S2). These observations suggest that HIWL disrupts the xanthosome transport system in L. crocea, but this disruption is recoverable upon returning to LIWL conditions.

Effect of high-intensity white light on ventral skin color in L. crocea Effect of LIWL and HIWL on ventral skin color (A–D). (A) Fish kept in darkness were exposed to LIWL for 5 min, followed by continued exposure to LIWL for 40 min; (B) Fish kept in darkness were exposed to LIWL for 5 min, followed by exposure to HIWL for 40 min; (C) Magnified view of the red boxed area in (B). (D) Morphology of xanthophores in the red boxed area of (C). Duration of the effect of HIWL on ventral skin color (E and F). (E) Fish previously kept in darkness were first exposed to LIWL for 5 min, followed by HIWL for 40 min, and subsequently returned to LIWL for an additional 4 h; (F) Morphology of xanthophores in ventral skin shown in (E). HIWL: high-intensity white light, 10,000 lux, 3.86 mW/cm2; LIWL: low-intensity white light, 150 lux, 0.04 mW/cm2; Scale bar: 50 μm.

Effect of different light intensities of white light on ventral skin color in L. crocea (A) The color and (B) morphology of xanthophores in ventral skins exposed to white light with different light intensities of 5,000 lux (1.93 mW/cm2), 8,000 lux (3.09 mW/cm2), and 10,000 lux (3.86 mW/cm2) for 40 min. (C) Statistical analysis of the proportion of xanthophores with dispersed xanthosomes in (B) (n = 3 independent experiments). Statistical significance was determined by t-test analysis. Scale bar: 50 μm.
Xanthosomes dispersion induced by high-intensity light is independent of light spectrum
Previous studies have shown that several photoreceptor genes are expressed in the xanthophores of L. crocea 15. We hypothesized that xanthosome dispersion triggered by HIWL illumination might be mediated by a photoreceptor sensitive to a specific wavelength of within the light spectrum. To test this hypothesis, xanthophores were initially exposed to LIWL to induce xanthosome aggregation, followed by exposure to high-intensity blue light (10,000 lux, 7.11 mW/cm2, λmax = 480 nm) or high-intensity red light (10,000 lux, 22.20 mW/cm2, λmax = 686 nm) (Fig. S1). The results showed that both blue and red light induced xanthosome dispersion (Fig. 3), suggesting that the dispersion caused by HIWL illumination is not mediated by photoreceptors responsive to specific wavelengths. Additionally, the baseline temperature of the cell culture medium was equivalent to the ambient room temperature on the experimental day, and no statistically significant temperature increases were detected in the HIWL, high-intensity blue light, and high-intensity red light treatment groups (Supplemental Fig. S3). These findings indicate that the mechanism behind xanthosome dispersion under HIWL operates independently of photoreceptor activation and temperature.

Effects of different wavelengths of light on xanthosomes movement Ex vivo effect of LIWL (A), high-intensity blue light (B) and high-intensity red light (C) on xanthosomes movement (n = 3 independent experiments). (D) Statistical analysis of the proportion of xanthophores with dispersed xanthosomes in experimental groups illuminated with different light sources. Statistical significance was determined by t-test analysis. High-intensity blue light λmax = 480 nm, 7.11 mW/cm2; High-intensity red light λmax = 686 nm, 22.20 mW/cm2. Red arrows indicate dispersed xanthosomes; LIWL: low-intensity white light, 150 lux, 0.04 mW/cm2; Scale bar: 50 μm.
PKA & PKC signaling pathways were not involved in xanthosomes dispersion by HIWL
Our previous study demonstrated that xanthosome dispersion is mediated via the cyclic adenosine monophosphate-protein kinase A (cAMP-PKA) signaling pathway12. To determine whether xanthosome dispersion induced by HIWL was due to the activation of PKA, we treated xanthophores under ex vivo culture conditions with 50 μM H-89 2HCl, a PKA pathway inhibitor, and exposed them to LIWL for 1 h, followed by HIWL for 40 min (Fig. 4A). The results showed that inhibition of the PKA pathway did not block xanthosome dispersion induced by HIWL illumination (Fig. 4B). According to previous studies, light-induced xanthosome aggregation is mediated via the Ca2⁺-protein kinase C (Ca2⁺-PKC) signaling pathway15. To investigate whether HIWL-induced xanthosome dispersion is caused by inhibition of the Ca2⁺-PKC signaling pathway, we treated xanthophores with 100 μM phorbol 12-myristate 13-acetate (PMA), a PKC activator, and exposed them to HIWL for 40 min, followed by either LIWL or HIWL. In the LIWL group, xanthosomes aggregated 3 h after dark treatment (Fig. 4C). Conversely, in the HIWL group, xanthosomes remained dispersed after exposure (Fig. 4D). These findings indicate that neither the activation of the cAMP-PKA pathway nor the inhibition of the Ca2⁺-PKC pathway is involved in the dispersion of xanthosomes induced by HIWL.

Effect of H-89 2HCl and PMA on xanthosomes dispersion under HIWL Ex vivo effect of blocking the cAMP-PKA pathway on xanthosome dispersion under HIWL (A and B). (A) The xanthophores were treated with H-89 2HCl for 1 h in LIWL condition, followed by HIWL treatment for 40 min. DMSO was used as the control (n = 3 independent experiments). (B) Statistical analysis of the proportion of xanthophores with dispersed xanthosomes in (A). Statistical significance was determined by t-test analysis. Ex vivo effect of activating the Ca2⁺-PKC pathway on xanthosome dispersion under HIWL (C and D). (C) The xanthophores were treated with PMA in LIWL condition, followed by HIWL treatment for 40 min, and followed by dark /HIWL treatment for 3 h (n = 3 independent experiments). (D) Statistical analysis of the proportion of xanthophores with dispersed xanthosomes at 0, 40 min, and 3 h 40 min in both groups. Statistical significance was determined by two‐way ANOVA analysis. Red arrows indicate dispersed xanthosomes; HIWL: high-intensity white light, 10,000 lux, 3.86 mW/cm2; LIWL: low-intensity white light, 150 lux, 0.04 mW/cm2; Scale bar: 50 μm.
HIWL-induced xanthosomes dispersion was mediated by microtubule depolymerization
The movement of pigment granules within chromatophores is facilitated by the cytoskeleton, primarily composed of microfilaments and microtubules37. In our previous study, we demonstrated that xanthosome movement is largely dependent on microtubules15 and xanthosome dispersion is dependent on both microtubules and microfilaments38. To investigate whether microtubules are involved in HIWL-induced xanthosome dispersion, we treated xanthophores with Paclitaxel (PTX), a microtubule-stabilizing agent. After 1 h of LIWL illumination and followed by 40 min of HIWL exposure, xanthosomes treated with 250 nM PTX remained aggregated (Fig. 5A and B). Additionally, xanthophores treated with 1 μM Nocodazole, a known microtubule depolymerizer39, and 50 μM H-89 2HCl (a PKA inhibitor), displayed xanthosome dispersion even under LIWL conditions (Fig. 5C and D). These results suggest that xanthosome dispersion under HIWL is likely mediated by microtubule depolymerization.

HIWL induced xanthosomes dispersion by depolymerizing microtubules. Ex vivo effect of Paclitaxel on xanthosome dispersion under HIWL (A). The xanthophores were treated with paclitaxel in LIWL condition for 1 h, followed by HIWL treatment for 40 min. DMSO was used as the control (n = 3 independent experiments). (B) Statistical analysis of the proportion of xanthophores with dispersed xanthosomes across different Paclitaxel concentrations under HIWL. Statistical significance was determined by one‐way ANOVA analysis. Ex vivo effect of blocking the PKA pathway and inhibiting microtubules on xanthosome movement (C). The xanthophores were treated with H 89 2HCI or DMSO in LIWL condition for 1 h, followed by co-treated with H 89 2HCI or DMSO and nocodazole in LIWL condition for 40 min, DMSO was used as the control (n = 3 independent experiments). (D) Statistical analysis of the proportion of xanthophores with dispersed xanthosomes in (C). Statistical significance was determined by t-test analysis. Red arrows indicate dispersed xanthosomes; PTX: Paclitaxel; NOC: Nocodazole; HIWL: high-intensity white light, 10,000 lux, 3.86 mW/cm2; LIWL: low-intensity white light, 150 lux, 0.04 mW/cm2; Scale bar: 50 μm.
HIWL inhibited microtubule assembly by increasing intracellular Ca2+
Intracellular Ca2⁺ plays a crucial role in maintaining microtubule stability40,41,42,43,44. To mimic the increase in intracellular Ca2⁺ levels, we treated xanthophores with A23187, a cell membrane-permeable Ca2⁺ ionophore. The results showed that 100 μM A23187 induced xanthosome dispersion in xanthophores (Fig. 6A, B). The ryanodine receptor (RyR) and inositol 1,4,5-trisphosphate receptor (IP₃R) are well-established Ca2⁺ release channels within ER. To investigate the role of IP₃R in this process, we treated xanthophores with 100 μM 2-aminoethyl diphenylborinate (2-APB), a membrane-permeable IP₃R inhibitor, for 30 min, followed by HIWL illumination for 1 h. Under these conditions, xanthosomes did not disperse (Fig. 6C, D). Taken together, these results suggest that HIWL triggers the release of Ca2⁺ through IP₃R channels in the ER, leading to an increase in intracellular Ca2⁺ levels and subsequent microtubule depolymerization.

HIWL induced upregulation of intracellular Ca2+ in xanthophore by activating IP3R Ca2+ channel of endoplasmic reticulum. (A) Ex vivo effects of 100 μM A23187 on xanthosomes movement under LIWL. The xanthophores were first culture under LIWL condition, followed by treatment with A23187 for 30 min. DMSO was used as the control (n = 3 independent experiments). (B) Statistical analysis of the proportion of xanthophores with aggregated xanthosomes in groups treated with different concentration of A23187. Statistical significance was determined by one‐way ANOVA analysis. (C) Ex vivo effects of 100 μM 2-APB on xanthosomes movement under HIWL. The xanthophores were treated with 2-APB in LIWL condition for 30 min, followed by HIWL treatment for 1 h. DMSO was used as the control (n = 3 independent experiments). (D) Statistical analysis of the proportion of xanthophores with aggregated xanthosomes in group treated with 100 μM 2-APB. Statistical significance was determined by t-test analysis. Red arrows indicate dispersed xanthosomes; HIWL: high-intensity white light, 10,000 lux, 3.86 mW/cm2; LIWL: low-intensity white light, 150 lux, 0.04 mW/cm2; Scale bar: 50 μm.
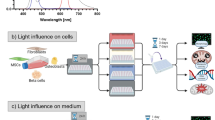
HIWL induced microtubule depolymerization in eukaryotic cells
Microtubules are tubular structures commonly found in eukaryotic cells. To determine whether HIWL induces microtubule depolymerization in other cell types, we extended our investigation to HeLa and HEK293T cells. HIWL resulted in an upregulation of intracellular Ca2⁺ in HeLa cells (Fig. 7A, B), which could be inhibited by 2-APB (Fig. 7C, D). We examined the morphology of microtubules in HeLa cells using immunohistochemistry with an anti-tubulin antibody. Under LIWL, the microtubules in HeLa cells were evenly distributed; however, after 40 min of HIWL exposure, the microtubules collapsed, losing their characteristic tubular structure (Fig. 7E). Further, treatment of HeLa cells with 10 μM A23187, a Ca2⁺ ionophore, induced microtubule depolymerization (Fig. 7F). In contrast, treatment with 10 μM BAPTA-AM, a Ca2⁺ chelator, inhibited this depolymerization (Fig. 7G). Similarly, 100 μM 2-APB inhibited microtubule depolymerization caused by HIWL exposure in HeLa cells (Fig. 7H). Similar to HeLa cells, HIWL exposure can also induce intracellular Ca2⁺ elevation and microtubule depolymerization in HEK293T cells, and this phenomenon can be blocked by BAPTA-AM through chelating intracellular Ca2⁺ (Supplemental Fig. S4). Taken together, these results suggest that HIWL triggers the release of Ca2⁺ through IP₃R in ER, leading to an increase in intracellular Ca2⁺ and subsequent microtubule depolymerization (Fig. 8). These findings indicate that HIWL-induced microtubule depolymerization is a widespread phenomenon across different eukaryotic cells.

HIWL induced microtubule depolymerization in HeLa cells. (A) Image and (B) signal intensity of Fluo-4 fluorescence in HeLa cells after 40 min of LIWL or HIWL exposure (n = 3 independent experiments). Statistical significance was determined by t-test analysis. (C) Image and (D) signal intensity of Fluo-4 fluorescence in in HeLa cells treated with DMSO or 2-APB under HIWL condition (n = 3 independent experiments). Statistical significance was determined by t-test analysis. (E) Effect of HIWL on morphology of microtubules in HeLa cells. (F) Effect of the A23187 on microtubules in HeLa cells. (G) Effect of BAPTA-AM on microtubules in HeLa cells under HIWL. (H) Effects of 2-APB on microtubules in HeLa cells under HIWL. Fluo-4 (green): Ca2+ fluorescence signal; DAPI (blue): Cell nuclei; Tubulin (green); Merge: Merged image of DAPI and Tubulin; Red arrows indicate microtubule depolymerization; HIWL: high-intensity white light, 10,000 lux, 3.86 mW/cm2; LIWL: low-intensity white light, 150 lux, 0.04 mW/cm2; Scale bar: 100 μm (A, C); 10 μm (E–H).

The molecular mechanism of xanthosomes dispersion caused by high-intensity light illumination in L. crocea. (A) High-intensity light triggered the release of Ca2+ through IP3R calcium ion channels in the ER of xanthophores. (B) Sustained Ca2⁺ release through IP₃R channels led to elevated intracellular Ca2⁺ levels, which subsequently induced microtubule depolymerization. (C) Microtubule depolymerization disrupts centripetal transport along microtubule tracks, leading to xanthosome dispersion.
Discussion
In recent years, excessive intensities of light exposure caused cell dysfunction, e.g., altered gene expression, reduced cell motility, diminished cell viability and mitosis as well as increased apoptosis, have attracted significant attention45,46,47,48,49. In this study, we found that the xanthosomes of large yellow croaker dispersed under HIWL exposure, which was significantly different from xanthosomes aggregation observed under LIWL conditions. We further revealed that prolonged HIWL exposure activates IP3R calcium ion channels on ER, leading to an acute increase in intracellular Ca2+ concentration and subsequent microtubule depolymerization. This disruption severely impairs the microtubule-based intracellular transport network in xanthophores, resulting in xanthosomes dispersion. Our findings deepen our understanding of the critical role of the cytoskeleton in the intracellular transport network of pigment granules and other substances, while also highlighting the detrimental effects of HIWL exposure on cellular function.
In teleostean chromatophores, cAMP, Ca2⁺, and IP₃ have been identified as key second messengers. A decrease in cAMP concentration induces pigment aggregation in chromatophores50,51, while an increase in Ca2⁺52,53,54 or IP₃55 levels triggers a similar response. Our previous study demonstrated that Opsin 3, a blue light-sensitive photoreceptor, localized in xanthophores, mediates LIWL-dependent intracellular Ca2⁺ elevation, consequently triggering activation of the Ca2⁺-PKC signaling cascade15. This pathway then acts on dynein, promotes centripetal microtubule movement, and facilitates xanthophore aggregation. This process is independent of the visual system15. In this study, high-intensity blue light and red light were all capable of inducing xanthosome dispersion in isolated xanthophores. The inhibition of xanthosome movement-associated signaling pathways failed to prevent HIWL-induced xanthosome dispersion. These results demonstrate that HIWL-induced xanthosome dispersion is independent of visual system and photoreceptors in xanthophore.
Considering the dispersion of xanthosomes in response to HIWL is unrelated to the Opsin 3-signaling pathway, we hypothesize the xanthosomes dispersion is related to cytoskeleton. Carlton et al. has shown that light exposure can inhibit yeast cell activity by suppressing microtubules56. Pharmacological experiments targeting the cytoskeleton support this conclusion. Treatment with the microtubule-stabilizing agent paclitaxel, which prevents microtubule depolymerization without affecting their assembly57, effectively inhibited xanthosome dispersion under HIWL illumination. The cAMP-PKA signaling pathway has long been implicated in providing centrifugal motility for pigment granules50,51. However, even when the PKA pathway is inhibited and microtubules within xanthophores are depolymerized, xanthosome dispersion still occurs. This observation suggests that the loss of microtubules as tracks for xanthosome aggregation is accompanied by a concurrent loss of the PKA signaling pathway, which normally provides the dispersal force for these granules. Our previous study indicated that xanthosomes dispersion is partially relied on the actin15,38, which may be the only reason why xanthosome can still disperse even after detaching from the microtubules. In this study, the significant inhibition of microtubule stability in ex vivo xanthophores effectively suppressed the HIWL-induced xanthosome dispersion, suggesting that the dispersion of xanthosomes may result from the microtubule depolymerization caused by HIWL.
Earlier studies on the requirements for in vitro microtubule polymerization have shown that Ca2⁺ is an effective inhibitor of microtubule formation58,59,60,61,62. To investigate whether microtubule depolymerization in xanthophores, resulting in xanthosome dispersion under HIWL, is due to increased intracellular Ca2⁺, we initially considered using calcium spark assays. However, this approach proved impractical, as capturing weak calcium spark signals under 10,000 lux of HIWL was not feasible. In this study, exogenous supplementation of excessive intracellular Ca2⁺ using ionophores A23187 induced microtubule depolymerization in xanthophores. Besides, the Fluo-4 Ca2⁺ indicator experiment in HeLa cells showed that exposure to HIWL significantly upregulated intracellular Ca2⁺. These results supported the hypothesis that elevated Ca2⁺ levels contribute to microtubule depolymerization. Based on these findings, we propose that HIWL induces acute upregulation of intracellular Ca2⁺, leading to microtubule depolymerization. The concentration of Ca2⁺ required to effectively inhibit microtubule assembly in vitro has been reported with considerable variation across studies. Early observations by Weisenberg suggested inhibition at a concentration as low as 6 μM40. In contrast, Lee and Haga reported an effective inhibitory concentration of around 30 μM59,60, while Olmsted observed microtubule inhibition only at concentrations exceeding 1 mM63. Schliwa also noted that a Ca2⁺ concentration of 10 μM had a degrading effect on the axopodia of heliozoans64. In this study, treatment with A2318765 at concentrations as low as 10 μM significantly inhibited microtubule polymerization in HeLa cells. However, in primary xanthophores, a higher concentration of 100 μM was required to induce xanthosome dispersion. This discrepancy likely reflects differences in cellular sensitivity to ionophores, as well as variations in the rate of Ca2⁺ influx across different cell types. In conclusion, these findings provide strong support for our hypothesis that HIWL illumination elevates intracellular Ca2⁺ levels, leading to microtubule depolymerization and subsequent xanthosome dispersion.
In resting cells, intracellular free Ca2⁺ concentrations are tightly regulated and maintained at significantly lower levels than those in the extracellular environment. Specifically, there exists a 20,000-fold gradient between the extracellular fluid, which contains approximately 1.2 mM of Ca2⁺, and the intracellular cytoplasm, where concentrations range from 10 to 100 nM. Within the endoplasmic reticulum (ER), the primary intracellular Ca2⁺ store, Ca2⁺ concentrations range between 100 and 800 µM66. Under HIWL illumination, intracellular Ca2⁺ increase is likely attributed to the release of Ca2⁺ from these internal stores. The two primary Ca2⁺ release channels in the ER are the RyR and IP₃R. Since RyR is not active in HeLa cells67,68,69, we focused our study on the IP₃R Ca2⁺ channel70,71. IP₃Rs are large-conductance cation channels that are activated in response to the stimulation of cell surface receptors72. In the original study by Maruyama et al., it was shown that 2-APB (2-aminoethoxydiphenyl borate) can affect multiple processes involved in cellular Ca2⁺ homeostasis34. 2-APB has been widely used to demonstrate the role of IP₃Rs in intracellular signaling, and it has also been shown to block store-operated Ca2⁺ channels (SOC)33. Peppiatt et al. reported that 2-APB can rapidly cross the plasma membrane of HeLa cells, completely inhibiting Ca2⁺ mobilization evoked by inositol 1,4,5-trisphosphate (InsP₃) esters. They further demonstrated that 100 μM 2-APB effectively inhibited histamine- and ATP-evoked Ca2⁺ signals35. In this study, we observed that 100 μM 2-APB completely blocked the microtubule depolymerization in HeLa and HEK293T cells exposed to HIWL illumination. A similar effect was noted in xanthophores, where inhibition of the IP₃R Ca2⁺ channels prevented xanthosome dispersion under the same light conditions. These findings suggest that the rapid increase in intracellular Ca2⁺ originates from the ER via IP₃R-mediated release. Several studies have demonstrated that excessive light exposure elevates intracellular reactive oxygen species (ROS) levels in mammalian cells, thereby inducing endoplasmic reticulum (ER) stress and IP₃R-mediated Ca2⁺ release from the ER, which subsequently results in a significant increase in intracellular Ca2⁺ levels16,17,18,19. Further study would be necessary to determine whether HIWL exposure induced Ca2⁺ releasement from IP3R channel was due to ER stress response.
In summary, HIWL targets the IP3R calcium channel, inducing a significant increase in intracellular Ca2+ concentration in xanthophores. This surge in Ca2⁺ disrupts the microtubule assembly process, destabilizing the dynamic equilibrium of microtubule complexes and ultimately leading to microtubule depolymerization. Consequently, xanthosomes lose their microtubule transport tracks, resulting in their dispersion. Our findings provide a research model for understanding how HIWL exposure affects intracellular organelle dynamics and cellular transport networks. Furthermore, the discovery that HIWL-induces microtubule depolymerization provides mechanistic insights into its cytotoxic effects, thereby proposing a novel strategy for suppressing tumor cell proliferation through photodynamic cytoskeletal modulation.
Data availability
The data that support this study are available from the corresponding author upon request.
References
Valiron, O., Caudron, N. & Job, D. Microtubule dynamics. Cell. Mol. Life Sci. CMLS 58, 2069–2084 (2001).
Nascimento, A. A., Roland, J. T. & Gelfand, V. I. Pigment cells: a model for the study of organelle transport. Annu. Rev. Cell Dev. Biol. 19, 469 (2003).
Bikle, D., Tilney, L. & Porter, K. Microtubules and pigment migration in the melanophores of Fundulus heteroclitus L. Protoplasma 61(3), 322–345 (1966).
Rodionov, V. I., Gyoeva, F. K. & Gelfand, V. I. Kinesin is responsible for centrifugal movement of pigment granules in melanophores. Proc. Natl. Acad. Sci. 88(11), 4956–4960 (1991).
Rodionov, V. et al. Switching between microtubule-and actin-based transport systems in melanophores is controlled by cAMP levels. Curr. Biol. 13(21), 1837–1847 (2003).
Nilsson, H. & Wallin, M. Evidence for several roles of dynein in pigment transport in melanophores. Cell Motil. Cytoskelet. 38(4), 397–409 (1997).
Tuma, M. C. & Gelfand, V. I. Molecular mechanisms of pigment transport in melanophores. Pigment Cell Res. 12(5), 283–294 (1999).
Porta, G. D. & Mühlbock, O. Structure and control of the melanocyte. Quart. Rev. Biol. https://doi.org/10.1007/978-3-642-99906-2 (1967).
Palazzo, R. E. et al. Rearrangements of pterinosomes and cytoskeleton accompanying pigment dispersion in goldfish xanthophores. Cell Motil. Cytoskelet. 13(1), 9–20 (1989).
Tchen, T. et al. Role of microtubules in the formation of carotenoid droplet aggregate in goldfish xanthophores a. Ann. N. Y. Acad. Sci. 466(1), 887–894 (1986).
Chen, S., Su, Y. & Hong, W. Aquaculture of the large yellow croaker. Aquacult. China 10, 297–308 (2018).
Han, J. et al. The regulation of melanocyte-stimulating hormone on the pigment granule dispersion in the xanthophores and melanophores of the large yellow croaker (Larimichthys crocea). Aquaculture 507, 7–20 (2019).
Shi, C. & Chen, S. X. Structural and ultrastructural aspects of the skin of large yellow croaker Larimichthys crocea. J. Fish Biol. https://doi.org/10.1111/jfb.15718 (2024).
Shi, C. et al. Protocol for isolating pigment cells from skin of large yellow croaker larimichthys crocea (Sciaenidae). J. Ichthyol. https://doi.org/10.1134/S0032945224700838 (2025).
Zhang, Z. et al. Nonvisual system-mediated body color change in fish reveals nonvisual function of Opsin 3 in skin. J. Photochem. Photobiol., B 252, 112861 (2024).
Nakanishi, T. et al. Role of endoplasmic reticulum stress in light-induced photoreceptor degeneration in mice. J. Neurochem. 125(1), 111–124 (2013).
Yang, L. P. et al. Endoplasmic reticulum stress is activated in light-induced retinal degeneration. J. Neurosci. Res. 86(4), 910–919 (2008).
Shah, S. Z. A. et al. Regulatory mechanisms of endoplasmic reticulum resident IP3 receptors. J. Mol. Neurosci. https://doi.org/10.1007/s12031-015-0551-4 (2015).
Kiviluoto, S. et al. Regulation of inositol 1, 4, 5-trisphosphate receptors during endoplasmic reticulum stress. Biochimica et Biophys Acta (BBA) Mol Cell Res 183(7), 1612–1624 (2013).
Rakers, S. et al. Pros and cons of fish skin cells in culture: long-term full skin and short-term scale cell culture from rainbow trout, Oncorhynchus mykiss. Eur. J. Cell Biol. 90(12), 1041–1051 (2011).
Tan, F. et al. Comparative evaluation of the cytotoxicity sensitivity of six fish cell lines to four heavy metals in vitro. Toxicol. In Vitro 22(1), 164–170 (2008).
Ballester, R. & Rosen, O. M. Fate of immunoprecipitable protein kinase C in GH3 cells treated with phorbol 12-myristate 13-acetate. J. Biol. Chem. 260(28), 15194–15199 (1985).
Frederick, H. et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell 27, 397–408 (2015).
Jin, Z. et al. Protein kinase C-α (PKCα) modulates cell apoptosis by stimulating nuclear translocation of NF-kappa-B p65 in urothelial cell carcinoma of the bladder. BMC Cancer 17(1), 432–466 (2017).
Chijiwa, T. et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino) ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J. Biol. Chem. 265(9), 5267–5272 (1990).
Lee, T. H. & Linstedt, A. D. Potential role for protein kinases in regulation of bidirectional endoplasmic reticulum-to-Golgi transport revealed by protein kinase inhibitor H89. Mol. Biol. Cell 11(8), 2577–2590 (2000).
Wie, M. B. et al. BAPTA/AM, an intracellular calcium chelator, induces delayed necrosis by lipoxygenase-mediated free radicals in mouse cortical cultures. Prog. Neuropsychopharmacol. Biol. Psychiatry 25(8), 1641–1659 (2001).
Luyten, T. et al. Resveratrol-induced autophagy is dependent on IP3Rs and on cytosolic Ca2+. Biochimica et Biophysica Acta BBA Mol Cell Res 184(6), 947–956 (2017).
Thaler, C. D. & Haimo, L. T. Regulation of organelle transport in melanophores by calcineurin. J. Cell Biol. 111(5), 1939–1948 (1990).
Müller-Deku, A. et al. Photoswitchable paclitaxel-based microtubule stabilisers allow optical control over the microtubule cytoskeleton. Nat. Commun. 11(1), 1–12 (2020).
Leung, J. C. & Cassimeris, L. Reorganization of paclitaxel-stabilized microtubule arrays at mitotic entry: Roles of depolymerizing kinesins and severing proteins. Canc Biol. Ther. 20(10), 1337–1347 (2019).
Sack, U. et al. S100A4-induced cell motility and metastasis is restricted by the Wnt/β-catenin pathway inhibitor calcimycin in colon cancer cells. Mol. Biol. Cell 22(18), 3277–3553 (2011).
Bootman, M. D. et al. 2-Aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 16(10), 1145–1150 (2002).
Maruyama, T. et al. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins (1, 4, 5) P3-induced Ca2+ release. J. Biochem. 122(3), 498–505 (1997).
Peppiatt, C. M. et al. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1, 4, 5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium 34(1), 97–108 (2003).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9(7), 671–675 (2012).
Skld, H. N. et al. Fish chromatophores—From molecular motors to animal behavior. Int. Rev. Cell Mol. Biol. 321, 171–219 (2016).
Ge, X., The Study on the Function of Microfilaments, Microtubules and Their Motor Proteins in the Movement of Pigment Granules of Xanthophores in Large Yellow Croaker (in Chinese) Master Thesis, Xiamen University (2021).
Vasquez, R. J. et al. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol. Biol. Cell 8(6), 973–985 (1997).
Weisenberg, R. C. Microtubule formation in vitro in solutions containing low calcium concentrations. Science 177(4054), 1104–1105 (1972).
Fuller, G. et al. Involvement of mitochondria and calcium in regulating microtubule assembly in vitro. J. Cell Biol. 67(22), 126 (1975).
Bryan, J. Some factors involved in the control of microtubule assembly in sea urchins. Am. Zool. 15(3), 649–660 (1975).
Gallin, I. J. The regulatory role of divalent cations in human granulocyte chemotaxis: evidence for an association between calcium exchanges and microtubule assembly. J. Cell Biol. 62(3), 594–609 (1974).
Schlaepfer, W. W. Effects of calcium ion concentration on the degeneration of amputated axons in tissue culture. J. Cell Biol. https://doi.org/10.1083/jcb.59.2.456 (1973).
Icha, J. et al. Phototoxicity in live fluorescence microscopy, and how to avoid it. BioEssays 39(8), 1700003 (2017).
Dani, S. et al. The effect of continuous long-term illumination with visible light in different spectral ranges on mammalian cells. Sci. Rep. 14(1), 9444 (2024).
Wäldchen, S. et al. Light-induced cell damage in live-cell super-resolution microscopy. Sci. Rep. 5(1), 15348 (2015).
Alghamdi, R. A. et al. Assessing phototoxicity in a mammalian cell line: How low levels of blue light affect motility in PC3 cells. Front. Cell Dev Biol 9, 738786 (2021).
Schneckenburger, H. et al. Light exposure and cell viability in fluorescence microscopy. J. Microsc. 245(3), 311–318 (2012).
Novales, R. & Fujii, R. A melanin-dispersing effect of cyclic adenosine monophosphate on Fundulus melanophores. J. Cell. Physiol. 75(1), 133–135 (1970).
Miyashita, Y. & Fujii, R. Receptor mechanisms in fish chromatophores—II Evidence for beta adrenoceptors mediating melonosome dispersion in guppy melanophores. Comparat. Biochem. Physiol. Part C: Comparat. Pharmacol. 51(2), 179–187 (1975).
Luby-Phelps, K. & Porter, K. R. The control of pigment migration in isolated erythrophores of Holocentrus ascensionis (Osbeck) II. The role of calcium. Cell 29(2), 441–450 (1982).
Negishi, S., The role of calcium and magnesium on pigment translocation in melanophores of Oryzias latipes. Pigment cell 1985: Biological, molecular and clinical aspects of pigmentation, p. 233–239 (1985).
Noriko, O. et al. Pigment aggregation is triggered by an increase in free calcium ions within fish chromatophores. Comp. Biochem. Physiol. A Physiol. 91(1), 27–32 (1988).
Fujii, R., Wakatabi, H. & Oshima, N. Inositol 1, 4, 5-trisphosphate signals the motile response of fish chromatophores I Aggregation of pigment in the tilapia melanophore. J. Exper. Zool. 259(1), 9–17 (1991).
Carlton, P. M. et al. Fast live simultaneous multiwavelength four-dimensional optical microscopy. Proc. Natl. Acad. Sci. 107(37), 16016–16022 (2010).
Spencer, C. M. & Faulds, D. Paclitaxel. Drugs 48(5), 794–847 (1994).
Gaskin, F., Cantor, C. R. & Shelanski, M. L. Biochemical studies on the in vitro assembly and disassembly of microtubules. Annals New York Acad. Sci. 253, 133–146 (2010).
Haga, T., Abe, T. & Kurokawa, M. Polymerization and depolymerization of microtubules in vitro as studied by flow birefringence. FEBS Lett. 39(3), 291–295 (1974).
Lee, Y. C. et al. The in vitro polymerization of tubulin from beef brain. J Neurobiol https://doi.org/10.1002/neu.480050404 (1974).
Shelanski, M. L. & Cantor, G. C. R. Microtubule assembly in the absence of added nucleotides. Proc. Natl. Acad. Sci. 70(3), 765–768 (1973).
Weisenberg, C. R. Microtubule Formation in vitro in solutions containing low calcium concentrations. Science 177(404), 1104–1105 (1972).
Olmsted, J. & Borisy, G. Ionic and nucleotide requirements for microtubule polymerization in vitro. Biochemistry 14(13), 2996–3005 (1975).
Schliwa, M. The role of divalent cations in the regulation of microtubule assembly In vivo studies on microtubules of the heliozoan axopodium using the ionophore A23187. J. Cell Biol. 70(3), 527–540 (1976).
Kenney, D. M. & Chao, F. C. Ionophore-induced disassembly of blood platelet microtubules: Effect of cyclic AMP and indomethacin. J. Cell. Physiol. 103(2), 289–298 (1980).
Raffaello, A. et al. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 41(12), 1035–1049 (2016).
Diarra, A. et al. Histamine-evoked Ca2+ oscillations in HeLa cells are sensitive to methylxanthines but insensitive to ryanodine. Pflugers Arch. 426, 129–138 (1994).
Zhu, D.-M. et al. Reversible phosphorylation as a controlling factor for sustaining calcium oscillations in HeLa cells: Involvement of calmodulin-dependent kinase II and a calyculin A-inhibitable phosphatase. Biochemistry 35(22), 7214–7223 (1996).
Bennett, D. L. et al. Expression and function of ryanodine receptors in nonexcitable cells (∗). J. Biol. Chem. 271(11), 6356–6362 (1996).
Berridge, M. J. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96(4), 1261–1296 (2016).
Vallese, F. et al. ER-mitochondria calcium transfer, organelle contacts and neurodegenerative diseases. Cal. Signal https://doi.org/10.1007/978-3-030-12457-1_29 (2020).
Gaspers, L. D. et al. Hormone-induced calcium oscillations depend on cross-coupling with inositol 1, 4, 5-trisphosphate oscillations. Cell Rep. 9(4), 1209–1218 (2014).
Acknowledgements
We thank Professor Kai Huang and Engineer Chunfeng Zhao from College of Physical Science and Technology, Xiamen University, as well as Assistant Engineer Chunyan Tian from the Future Display Technology Institute, Xiamen University, for their support with optical equipment. We thank Chenchen Shi (PhD candidate) and Xiaoyu Ge (MS) of the College of Ocean and Earth Sciences, Xiamen University, for their assistance in collecting samples.
Funding
This work was supported by National Key Research and Development Program of China (2022YFD2401001), National Natural Science Foundation of China (No. U22A20528), Key Project of Agriculture in Fujian Province of China (No. 2021N0001), Seed Industry Innovation and Industrialization Project of Fujian Province (2021FJSCZY01), Science and Technology Major Project of Fujian Province (2021NZ033016).
Author information
Authors and Affiliations
Contributions
Zihao Zhang and Shixi Chen wrote the main manuscript text and Zihao Zhang prepared Figs. 1–8. All authors reviewed the manuscript. Animal ethics review All animal experiments should comply with the ARRIVE guidelines. All experiment protocols were approved by the Institutional Animal Care and Use Committee of Xiamen University; IACUC: XMULAC20230011. Acknowledgement We thank Professor Kai Huang and Engineer Chunfeng Zhao from College of Physical Science and Technology, Xiamen University, as well as Assistant Engineer Chunyan Tian from the Future Display Technology Institute, Xiamen University, for their support with optical equipment. We thank Chenchen Shi (PhD candidate) and Xiaoyu Ge (MS) of the College of Ocean and Earth Sciences, Xiamen University, for their assistance in collecting samples. Conflict of interest We declare that there is no potential conflict of interest that represents a conflict of interest in connection with the work submitted.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All animal experiments should comply with the ARRIVE guidelines. All experiment protocols were approved by the Institutional Animal Care and Use Committee of Xiamen University; IACUC: XMULAC20230011.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Z., Han, J., Wei, K. et al. High-intensity light disrupts intracellular organelle dynamics via microtubule depolymerization. Sci Rep 15, 20888 (2025). https://doi.org/10.1038/s41598-025-04434-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-04434-1
