
Abstract
Human topoisomerase II (topoII) is a well-known and validated target for cancer treatment. We previously reported a first set of 6-amino-tetrahydroquinazoline derivatives as novel human topoII inhibitors. Here, we report on the expansion and this molecular scaffold and present 17 additional analogs centered on the tetrahydropyrido[4,3-d]pyrimidine heterocycle. Some of these compounds exhibit promising topoII inhibitory and antiproliferative activities. Compound 24 (ARN21929) shows good in vitro potency, with an IC50 of 4.5 ± 1.0 µM, excellent kinetic and thermodynamic solubility, and good metabolic stability. Our results indicate that this new chemical class of topoII inhibitors deserves further exploration and represents a promising starting point for developing novel and potentially safer topoII-targeted anticancer drugs.
Similar content being viewed by others

Introduction
The biological function of human topoisomerase II (topoII) enzymes is to modulate DNA topology and regulate vital processes like DNA replication, transcription, recombination, and chromosome segregation1,2,3,4,5. TopoII is also a validated target in the fight against cancer1,6,7 and several drugs targeting topoII are in clinical use1,2. Specifically, valuable examples of these anticancer drugs are represented by daunorubicin, doxorubicin, etoposide and teniposide. All these molecules are tetracyclic ring structures with a sugar moiety belonging to two main chemical classes: anthracyclines and epipodophyllotoxins (Fig. 1A). Notably, they all act as topoII poisons by trapping the covalent enzyme/DNA cleavage complex formed during DNA topology modifications6,7,8. As a consequence, topoII generates double-stranded breaks in the genome, which activate apoptotic mechanisms and result in cancer cell death. Although effective, this strategy is also responsible for severe unwanted side effects, including the development of myelosuppression, cardiotoxicity (notably with anthracyclines like doxorubicin), and the risk of therapy-related secondary leukemia in patients6,7,8,9,10 representing a medical issue that has renewed the interest in the drug discovery community to seek new and safer topoII-targeted drugs4,11,12,13.
In this context, our group has previously reported tetrahydroquinazoline-based small molecules that are potent topoII inhibitors and do not act as topoII poisons14,15,16,17,18,19,20,21,22. Notably, these compounds chemically differ from topoII poison, as they present a central bicyclic core substituted with different types of (hetero)aryl groups on C2 and C4 positions (Fig. 1A). Building on these findings, here we outline the development of new 17 (tetrahydro)pyridopyrimidine derivatives, describing the structure-activity relationship (SAR) study that elucidated the chemical features essential for the activity (Fig. 1B). Some of them are potent topoII inhibitors, have promising antiproliferative activity against five different human cancer cells and display favorable in vitro pharmacokinetic properties. These findings highlight the potential of these promising derivatives for developing novel anticancer agents.

(A) Examples of topoII poison inhibitors in clinical use; (B) Scaffolds explored for developing new topoII inhibitors21.
Results and discussion
Exploring the structure-activity relationship of new topoii inhibitors toward ARN21934
In our previous work, we developed a class of 14 tetrahydroquinazoline derivatives that allowed the identification of the low micromolar selective topoIIα inhibitor ARN21934 (1, IC50 = 2 ± 0.5 µM, Table 1) starting from the hit compound 2 (IC50 = 160 ± 20 µM, Table 1)21,22. Indeed the compounds also exhibited a favorable drug-like profile both in vivo and in vitro. In particular, the chemical structure of ARN21934 features a central bicyclic core consisting of a partially saturated quinazoline bearing a pyridine, an aniline, and an amino group in positions 2, 4, and 6, respectively (Fig. 1B).
In the present work, we aim to develop new topoisomerase inhibitors with a modified core heterocycle. During the previous SAR study, the unsubstituted tetrahydroquinazoline 3 (Table 1) confirmed that the presence of the exocyclic amino group in position 6 was essential for topoII inhibition. However, its stereochemistry did not influence the inhibitory activity, as shown by enantiopure compounds 4 and 5 (Table 1)21. Therefore, we designed two new scaffolds (Fig. 1B), shifting the nitrogen of the amino group in the endocyclic position and removing the chirality (Fig. 1). First, we replaced tetrahydroquinazoline with tetrahydropyrido[4,3-d]pyrimidine core as in compound 6. Thus, we mainly explored (i) pyridines with different positions of nitrogen and unsubstituted phenyl rings on C2, and (ii) anilines on position C4 with different substituents embedding electron-donating or electron-withdrawing groups in ortho, meta, and para positions, especially focusing on the alkylamino, methoxy, hydroxy, methyl and fluorine groups.
At this point, we also hypothesized that the substitution pattern in this new tetrahydropyridopyrimidine scaffold could establish interactions that favor topoII inhibition compared to the previously partially saturated quinazoline structures21. Therefore, we started a structure-activity relationship study (SAR) focusing our efforts on two points of modification: the aniline substituent on position 4 and the (hetero)aryl group on position 2 of the bicyclic core (Fig. 2; Table 1). First, we masked the free nitrogen of the aniline group on position 2 with a methyl group, synthesizing compound 9 (Table 1). As observed in previous studies, this modification was detrimental and resulted in a loss of activity against topoII, suggesting this NH group’s key hydrogen bond role21. With the second set of derivatives 10–12, we explored the importance of the 4-pyridyl substituent present in compound 7, replacing it with a phenyl (compound 10) or changing the anchor point of the pyridyl substituent into positions 3 and 2 (compounds 11 and 12, respectively). Differently from the previous tetrahydroquinazoline-based inhibitors21no one of the modifications was tolerated, and compounds 10–12 were inactive. After confirming that the 4-pyridyl heteroaromatic group was the best substituent in position 2 of the new tetrahydropyridopyrimidine scaffold and the essential role of the free nitrogen of aniline in position 4, we evaluated the decoration of the latter with analogs 13–24. First, introducing a naked phenyl group, as in compound 13, resulted in a loss of activity. Then, we explored groups in para- and meta- positions with different electronic and steric hindrance properties, such as fluorine (14), methyl (15 and 16), hydroxyl (17 and 18), methoxyl (19 and 20), monomethylamino (21 and 22), and dimethylamino (23) groups. This set of analogs’ behavior differed from the series described in our previous work21. In our new tetrahydropyrido[4,3-d]pyrimidine inhibitors, none of the substituents explored in the aniline para- position successfully increased inhibitory activity against topoII. Only substituents containing methylated oxygen or nitrogen in the aniline meta- position, as in compounds 20, 22, and 23, restored the activity of the starting hit, displaying an IC50 of 120, 160, and 140 µM, respectively. From these results, we speculated that most likely due to steric hindrance factors, a fluorine atom was the best decoration for the aniline substituent. At this point, we moved it into ortho- position, resulting in the single-digit micromolar topoII inhibitor 24 (ARN21929, IC50 4.5 ± 1.0 µM), which was five-fold more potent than compound 7 and with comparable activity of the previous lead ARN21934. In addition to its minimal steric impact, fluorine in ortho position of ARN21929 may alter the basicity of the adjacent aniline nitrogen and heterocycle core, potentially contributing to the improved binding profile and activity observed in these derivatives.

SAR campaign to identify ARN21929 (24).
We also evaluated the antiproliferative effect of the active compounds 7, 20–24 in a small panel of cancer cells (Table 2). Indeed, derivatives 1, 7, and 20 exhibited good or moderate antiproliferative activity in the low micromolar range against melanoma cell line (A375), breast (MCF7), endometrial (HeLa), lung (A549), and androgen-independent prostate (DU145) cancer cell lines. On the other hand, despite its promising inhibitory activity, ARN21929 showed IC50 > 100 µM against the same cancer cell lines. This limited cellular response may be associated with low cell permeability of ARN21929 compared to other analogues. Nonetheless, we note that ARN19661 (compound 7), which differs from ARN21929 by the position of a single fluorine atom, exhibited antiproliferative activity similar to the previous lead compound ARN21934, with IC₅₀ values in the 20–30 µM range. This promising result indicates that even minimal chemical modifications to this tetrahydroquinazoline-based scaffold can improve the overall activity.
Finally, we performed preliminary in vitro studies to assess the drug-likeness of ARN21929. This compound presented optimal kinetic (> 250 µM) and thermodynamic (596 µM) solubility. In terms of its metabolic stability, ARN21929 was stable in mouse plasma (t1/2 > 120 min) while showing moderate half-life in mouse liver microsomes (t1/2 = 30 min). Thus, despite the suboptimal antiproliferative activity, ARN21929 represents a viable and promising starting point for further exploration to develop a novel class of drug-like Topo II inhibitors with improved potency and efficiency.
Molecular modeling
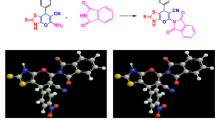
The binding mode of novel human topoisomerase II (TopoII) inhibitors based on a tetrahydropyrido[4,3-d]pyrimidine molecular scaffold was investigated through molecular docking23. To assess alternative and functionally relevant inhibitory mechanisms, docking simulations were conducted considering three distinct binding sites. The first site, corresponding to the DNA cleavage region, was modeled using PDB entry 5GWK, which represents the drug-stabilized cleavage complex bound to etoposide24. The second and third sites explored the ATP-competitive mechanism at the N-terminal ATP-binding pocket, using PDB entry 1ZXM8,25. In TopoII, the catalytic site contains one Mg²⁺ ion at the ATP-binding site, which is critical for stabilizing ATP or ADP within the pocket. Therefore, docking simulations were performed both in the presence and absence of the Mg²⁺ cation to account for the transiently inactive form of the enzyme21.
Docking results indicate that ARN21929 exhibits well-defined binding modes across all three target sites, each characterized by distinct interaction patterns. At the DNA cleavage site, the positively charged NH₂ group of ARN21929 forms a cation–π interaction with G’+3 and a hydrogen bond with its oxygen atom. The fluorine substituent interacts with the amino group of C–1, while van der Waals interactions with residues such as Met762, Met766, Gly642 and Arg487 further stabilize this binding mode. Additionally, the pyridine nitrogen in the para position adopts a geometrically favorable orientation that enables hydrogen bonding with the hydroxyl group at C–1, contributing to the overall stabilization of the ligand within the binding site (Fig. 3A). At the ATP-binding site of TopoII in the presence of Mg²⁺, the pyridyl group of ARN21929 forms a cation–π interaction with the metal ion and a hydrogen bond with Gly164, resembling the binding mode observed for the previous lead ARN21934 in the same site. Further stabilizing interactions include (i) one hydrogen bond between the positively charged NH₂ group of the tetrahydropyrido[4,3-d]pyrimidine scaffold and the side chain of Asn95; (ii) one hydrogen bond between a nitrogen atom in the pyrimidine ring and the side chain of Asn150; (iii) one hydrogen bond between the fluorine substituent and the side chain of Ile125; and (iv) and the interaction between the fluorine-substituted aniline ring and the side chain of Asn120. Additionally, favorable van der Waals interactions occur with surrounding binding pocket residues (Fig. 3B). In the absence of the Mg²⁺ cation, ARN21929 engages multiple stabilizing interactions within the ATP-binding site. These include (i) one hydrogen bond between the pyrimidine nitrogen and the side chain of Lys168; (ii) interactions between the pyrimidine ring and the backbone carbonyl oxygens of Thr147 and Ile141; (iii) a π–π stacking interaction with the aromatic ring of Phe142; (iv) interactions between the fluorine-substituted aniline and the side-chain oxygen of Asn120; (v) one hydrogen bond between the positively charged NH₂ group and the backbone oxygen of Asp94; (vi) and one additional hydrogen bond between the fluorine atom and the side chain amino group of Asn120. The binding is further stabilized by van der Waals interactions with nearby residues (Fig. 3C).
Overall, our docking studies at both the DNA cleavage and ATP-binding sites indicate that ARN21929 may inhibit TopoII by engaging distinct residues at different sites, suggesting a multifaceted mechanism of action (additional docking details in the Supporting Information).

Binding modes from docking calculations for ARN21929 at the DNA cleavage and ATP sites of TopoII. (A) Binding pose of ARN21929 within the DNA-cleaved complex. (B) Binding pose of ARN21929 within the ATP-binding site in the presence of Mg²⁺. (C) Binding pose of ARN21929 within the ATP-binding site in the absence of Mg²⁺. The protein and DNA (present only in A) are shown in semi-transparent cartoon representation, while the nucleotide bases and ligand are displayed in licorice representation. Residues involved in van der Waals interactions are shown in semi-transparent licorice representation. Details on the interactions of each compound in the pockets are reported in the main text.
Chemistry
The synthetic strategy presented in Scheme 1.a of Fig. 4 allowed obtaining most of the tested compounds (6–7, 10–11 and 13 − 24) through a five-step procedure. After forming the pyrimidone-like bicyclic heterocycle (intermediate A), positions 2 and 4 were activated by chlorination after refluxing in neat phosphorous oxychloride obtaining intermediate B with good yield of 71% after two steps. Then, position 4 was selectively functionalized with corresponding aniline derivatives taking advantage of its major reactivity towards aromatic nucleophilic substitution or palladium catalyzed reactions. Thus, we afforded intermediates C.1-C.14 (Table 3) from moderate to good yields. Then, 4-pyridyl, 3-pyridyl and phenyl substituents were introduced in position 2 of intermediates D.1-D.16 (Table 3) employing microwave-assisted Suzuki coupling. Final benzyl group removal under reductive conditions in the presence of Pd(OH)2-C as catalyst and ammonium formate as hydrogen source gave final desired compounds 6, 7, 10, 11, 13–24. Compounds 9 and 12 were synthesized with an alternative procedure (Fig. 4, Scheme 1.b). First, the bicyclic heterocycle of F.1 and F.2 was assembled directly functionalized in position 2, by reacting methyl 1-benzyl-4-oxo-piperidine-3-carboxylate with the corresponding pyridine-carboxamidine. In the second step, position 4 of intermediate of type F was activated after chlorination under reflux in neat phosphorous oxychloride, giving compounds of type G with 74% yield. Afterwards, suitable anilines were introduced via microwave-assisted Buchwald reaction affording H1 and H2 with 37% and 60% yield respectively. Finally, benzyl group was removed under reductive conditions equivalent to the ones employed in previous Scheme 1.a of Fig. 4. The synthesis of compound 8 was approached with a similar strategy as depicted in Scheme 1.c of Fig. 4. Initially, the fully aromatic bicyclic core was formed by reaction of 4-aminopyridine-3-carboxylic acid and pyridine-4-carboxamidine yielding intermediate J functionalized in position 2 with 47% yield. In this case, the activation of position 4 (intermediate K) was carried on by refluxing in the presence of POCl3 and PCl5 to finally get compound 8 using a microwave-assisted Buchwald coupling reaction.

Scheme of synthetic routes for the obtainment of final compounds.
Conclusions
In summary, we have evolved our partially saturated quinazoline topoII inhibitors into a new class based on a tetrahydropyridopyrimidine scaffold that, notably, is not chiral. These results provide new insight into the key chemical features crucial for the inhibitory activity against topoII and the drug-like profiles of this chemical scaffold. Compounds ARN19661 (7) and ARN21929 (24) act as potent topoII inhibitors. Focused SAR studies showed that only small substituents in the 4-aniline group were partially tolerated. Indeed, introducing a fluorine atom in ortho- position significantly increased topoII inhibition. Among the new 17 tetrahydropyridopyrimidine derivatives, compounds ARN19661 (7), 20, 22, and 23 inhibit cell proliferation in the low-moderate micromolar range on five cancer cell lines. Moreover, ARN21929 (24) showed high kinetic and thermodynamic solubility and favorable in vitro metabolic parameters, thus further supporting this new chemical class as a promising starting point for further studies to develop novel therapeutic agents that may lack the development of secondary leukemias associated with topoII poisons.
Methods
Chemistry
Chemistry general considerations
All the commercial available reagents and solvents were used as purchased from vendors without further purification. Dry solvents were purchased from Sigma-Aldrich. Automated column chromatography purifications were done using a Teledyne ISCO apparatus (CombiFlash® Rf) with pre-packed silica gel columns of different sizes (from 4 g up to 120 g) and mixtures of increasing polarity of cyclohexane and ethyl acetate (EtOAc), cyclohexane and tert-butylmethyl eter (TBME) or dicloromethane (DCM) and methanol (MeOH). NMR experiments were run on a Bruker Avance III 400 system (400.13 MHz for1H, and 100.62 MHz for13C), equipped with a BBI probe and Z-gradients. Spectra were acquired at 300 K, using deuterated dimethyl sulfoxide (DMSO–d6), deuterated methanol (CD3OD) or deuterated chloroform (CDCl3) as solvents. For1H-NMR, data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = double of doublets, t = triplet, q = quartet, m = multiplet), coupling constants (Hz) and integration. UPLC-MS analyses were run on a Waters ACQUITY UPLC-MS system consisting of a single quadrupole detector (SQD) mass spectrometer equipped with an electrospray ionization interface (ESI) and a photodiode array detector (PDA) from Waters Inc. (Milford, MA, USA). Electrospray ionization in positive and negative modes was applied in the mass scan range 110 − 650 Da. The PDA range was 210–400 nm. The analyses were performed on an ACQUITY UPLC BEH C18 column (50 × 2.1mmID, particle size 1.7 μm) with a VanGuard BEH C18 pre-column (5 × 2.1 mmID, particle size 1.7 μm). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN–H2O (95:5) at pH 5.0 (B). Two types of gradients were applied depending on the polarity of the compounds: gradient 1 (5–95% mobile phase B in 2.5 min) or gradient 2 (50–100% mobile phase B in 2.5 min). A quality control (QC) by UPLC-MS (UV at 215 nm) was performed for all compounds before testing (see supporting information). A 10 mM stock solution of the compound was prepared in DMSO-d6, diluted 20-fold with MeCN − H2O (1:1), and analyzed on a Waters ACQUITY UPLC-MS system as defined above. The analyses were run on an ACQUITY UPLC BEH C18 column (100 × 2.1 mmID, particle size 1.7 μm) with a VanGuard BEH C18 pre-column (5 × 2.1 mm ID, particle size 1.7 μm). The mobile phase was 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in MeCN − H2O (95:5) at pH 5 (B) with 0.5 mL/min as the flow rate. A linear gradient was applied: 10% B for 0.2 min, 10 − 90% B in 6.0 min, 90 − 100% in 0.1 min, 100% B for 0.7 min.
High-resolution mass spectrometry (HRMS) for accurate mass measurements was performed on a Sciex TripleTOF High-resolution LC-MS using a Waters UPLC ACQUITY chromatographic system (from Waters Inc., Milford, MA, USA) coupled to a TripleTOF 5600 + Mass Spectrometer (from Sciex, Warrington, UK) equipped with a DuoSpray Ion source. The analyses were run on an ACQUITY UPLC BEH C18 column (50 × 2.1 mmID, particle size 1.7 μm), using H2O + 0.1% HCOOH (A) and MeCN + 0.1% HCOOH as mobile phase.
All key compounds showed ≥ 95% UV purity at 215 nm as determined by UPLC-MS analysis. (See supporting information for synthetic details).
General procedure 1. Reactions B scheme 1.a and G scheme 1.b (Fig. 4). Pyrimidone fused ring clorination
A suspension of corresponding pyrimidone fused ring (1 mmol) in POCl3 (1.5 ml) was stirred at 120 °C under N2 atmosphere until total solution was observed (around 4 h). POCl3 was then evaporated at low pressure, resulting residue solved in dichloromethane (3 ml), poured onto ice cold NaHCO3 saturated solution (18 ml), aqueous pH adjusted to 7–8 with NaHCO3 (no gas evolution observed after addition), organic layer separated, dried over Na2SO4 and concentrated to dryness at low pressure. Resulting solid normal phase chromatography purification finally yielded titled compound.
General procedure 2. Reactions C scheme 1.a. And L scheme 1.c (Fig. 4). Substituted aniline introduction
A suspension of chlorinated pyrimidine fused ring (1 mmol), corresponding aniline (1.1 mmol) and diisopropylethylamine (5 mmol) in 2-propanol (2 ml) was stirred in a CEM® microwave apparatus at 100–160 °C (depending of corresponding aniline) until reaction completion or no crude evolution was observed. Then reaction crude was concentrated to dryness at low pressure, solved in dichloromethane (20 ml), extracted with NaHCO3 saturated solution (20 ml), dried over Na2SO4 and concentrated to dryness at low pressure. Final normal phase purification yielded title compound.
General procedure 3. Reaction D scheme 1.a (Fig. 4). Suzuki coupling reaction
A suspension of compound obtained from general method C (1 mmol), corresponding boronic acid (1.2 mmol), PdCl2(dppf) dichloromethane complex (0.1 mmol) and K2CO3 2 M solution (2 mmol) in 1,4-dioxane (10 ml) was stirred in a CEM ® microwave apparatus at 120 °C for 2 h. Resulting crude was portioned between dichloromethane (25 ml), NaHCO3 saturated solution (25 ml), the organic layer dried over Na2SO4 and concentrated to dryness at low pressure. Final normal phase purification yielded title compound.
General procedure 4. Reactions E scheme 1.a and I scheme 1.b (Fig. 4). Benzyl group removal
Under N2 atmosphere, a suspension of compound to be deprotected (1 mmol), ammonium formate (4 mmol), Pd(OH)2/C (20% of starting material weight) was stirred at reflux temperature until reaction completion. Catalyst was filtered off with trough a celite coarse patch and resulting filtrate concentrated to dryness at low pressure. Final normal phase purification yielded title compound.
Synthesis of compound 6. N4,N4-dimethyl-N1-[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]benzene-1,4-diamine
Step 1. Synthesis of 6-benzyl-1,5,7,8-tetrahydropyrido[4,3-d]pyrimidine-2,4-dione (intermediate A). A suspension of methyl 1-benzyl-4-oxo-3-piperidinecarboxylate hydrochloride (5 g, 16.7 mmol) in ethanol (76 ml), urea (5.077 g, 83.7 mmol) and sodium methoxide (4.110 g, 75.3 mmol) was stirred at reflux temperature for 16 h. Afterwards the reaction crude was concentrated to dryness at low pressure, resulting solid triturated in water (20 ml), ice cooled, pH adjusted to 8–9 with concentrated HCl and filtrated. Resulting solid was then rinsed with methanol (20 ml) and diethyl ether (20 ml) yielding title compound (3.778 g, yield 88%). Rt = 1.30 min (gradient 1); MS (ESI) m/z: 258.1 [M + H]+, [M + H]+ calculated for C14H16N3O2: 258.1. 1H NMR (400 MHz, DMSO-d6) δ 10.90 (s, 1 H), 10.75 (s, 1 H), 7.39–7.29 (m, 4 H), 7.29–7.21 (m, 1 H), 3.62 (s, 2 H), 3.00 (d, J = 1.8 Hz, 2 H), 2.62 (t, J = 5.7 Hz, 2 H), 2.46–2.38 (m, 2 H).
Step 2. Synthesis of 6-benzyl-2,4-dichloro-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidine (intermediate B). Title compound was obtained using intermediate A (4.130 g, 16.05 mmol) following the general procedure 1 previously described. Final normal phase purification (cyclohexane/AcOEt from 95/5 to 75/25) afforded pure title compound (3.825 g, yield 81%). Rt = 2.53 min (gradient 2); MS (ESI) m/z: 294.0/296.0/298.0 [M + H]+, [M + H]+ calculated for C14H14Cl2N3: 294.0/296.0/298.0 (see supporting information for MS spectra of intermediate B). 1H NMR (400 MHz, CDCl3) δ 7.50–7.31 (m, 5 H), 3.87 (s, 2 H), 3.72 (s, 2 H), 3.08 (s, 2 H), 2.91 (s, 2 H).
Step 3. N1-(6-benzyl-2-chloro-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl)-N4,N4-dimethyl-benzene-1,4-diamine (Intermediate C.1). Title compound was obtained using intermediate B (200 mg, 0.68 mmol) and 4-dimethylaminoaniline (107.2 mg, 0.75 mmol) following the general procedure 2 previously described at 120 °C for 16 h. Final normal phase purification (cyclohexane/AcOEt from 70/30 to 50/50) afforded pure title compound (214 mg, yield 80%). Rt = 1.38 min (gradient 2); MS (ESI) m/z: 393.2/395.2 [M + H]+, [M + H]+ calculated for C22H24ClN5: 393.2/395.2. 1H NMR (400 MHz, DMSO-d6) δ 8.57 (s, 1 H), 7.42–7.31 (m, 4 H), 7.30–7.26 (m, 1 H), 7.26–7.22 (m, 2 H), 6.76–6.66 (m, 2 H), 3.74 (s, 2 H), 3.45 (s, 2 H), 2.88 (s, 6 H), 2.77–2.60 (m, 4 H).
Step 4. Synthesis of N4-[6-benzyl-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl]-N1,N1-dimethyl-benzene-1,4-diamine (intermediate D.1). Title compound was obtained using intermediate C.1 (210 mg, 0.53 mmol) and pyridine-4-boronic acid (87.4 mg, 0.64 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 40/60 to 10/90) afforded pure title compound (137.8 mg, yield 61%). Rt = 1.47 min (gradient 2); MS (ESI) m/z: 437.3 [M + H]+, [M + H]+ calculated for C27H28N6: 437.2. 1H NMR (400 MHz, DMSO- d6) δ 8.70–8.61 (m, 2 H), 8.35 (s, 1 H), 8.12–8.03 (m, 2 H), 7.51–7.44 (m, 2 H), 7.44–7.39 (m, 2 H), 7.39–7.33 (m, 2 H), 7.33–7.25 (m, 1 H), 6.83–6.72 (m, 2 H), 3.78 (s, 2 H), 3.57 (s, 2 H), 2.90 (s, 6 H), 2.85–2.79 (m, 2 H), 2.79–2.72 (m, 2 H).
Step 5. Synthesis of N4,N4-dimethyl-N1-[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]benzene-1,4-diamine (compound 6). Title compound was obtained using intermediate D.1 (135 mg, 0.31 mmol) following the general procedure 4 previously described for 5 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 60/40) afforded pure title compound (80.3 mg, yield 75%). Rt = 1.55 min (gradient 1); MS (ESI) m/z: 347.2 [M + H]+, [M + H]+ calculated for C20H23N6: 347.2. HRMS (ESI) m/z: 347.1979 calcd for C20H23N6 [M + H]+; found m/z: 347.1984. 1H NMR (400 MHz, DMSO-d6) δ 8.78–8.56 (m, 2 H), 8.22 (s, 1 H), 8.17–8.00 (m, 2 H), 7.64–7.39 (m, 2 H), 6.87–6.70 (m, 2 H), 3.75 (s, 2 H), 3.01 (t, J = 5.8 Hz, 2 H), 2.90 (s, 6 H), 2.73–2.64 (t, J = 5.8 Hz, 2 H), 2.60 (s, 1 H). 13C NMR (101 MHz, DMSO-d6) δ 161.0 (Cq), 157.5 (Cq), 157.1 (Cq), 150.1 (CH), 147.1 (C), 145.5 (C), 128.9 (C), 123.7 (CH), 121.3 (CH), 113.1 (C), 112.4 (CH), 42.6 (CH2), 42.4 (CH2), 40.5 (CH3), 32.0 (CH2). QC purity (UV at 215 nm) 99.5%.
Synthesis of compound 7. N-(3-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-chloro-N-(3-fluorophenyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (Intermediate C.2). Title compound was obtained using intermediate B (140 mg, 0.48 mmol) and 3-fluoroaniline (0.06 ml, 0.57 mmol) following the general procedure 2 previously described at 100 °C for 72 h. Final normal phase purification (cyclohexane/TBME from 90/100 to 50/50) afforded pure title compound (73 mg, yield 42%). Rt = 2.85 min (gradient 2); MS (ESI) m/z: 369.1/371.1 [M + H]+, [M + H]+ calculated for C20H18ClFN4: 369.1/371.1. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (s, 1 H), 7.61–7.45 (m, 1 H), 7.44–7.31 (m, 7 H), 7.31–7.23 (m, 1 H), 6.99–6.88 (m, 1 H), 3.76 (s, 2 H), 3.54 (s, 2 H), 2.72 (s, 4 H).
Step 2. Synthesis of 6-benzyl-N-(3-fluorophenyl)-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate D.2). Title compound was obtained using intermediate C.2 (74 mg, 0.20 mmol) and pyridine-4-boronic acid (32.9 mg, 0.24 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 50/50 to 20/80) afforded pure title compound (65.2 mg, yield 79%). Rt = 2.96 min (gradient 2); MS (ESI) m/z: 412.2 [M + H]+, [M + H]+ calculated for C25H23FN5: 412.2. 1H NMR (400 MHz, DMSO- d6) δ 8.70 (dd, J = 4.4, 1.7 Hz, 3 H), 8.17–8.04 (m, 2 H), 7.67 (dt, J = 12.0, 2.3 Hz, 1 H), 7.56 (ddd, J = 8.3, 2.0, 0.9 Hz, 1 H), 7.45–7.39 (m, 3 H), 7.39–7.34 (m, 2 H), 7.32–7.25 (m, 1 H), 6.91 (tdd, J = 8.5, 2.6, 0.9 Hz, 1 H), 3.80 (s, 2 H), 3.65 (s, 2 H), 2.87 (t, J = 5.8 Hz, 2 H), 2.78 (t, J = 6.1 Hz, 2 H).
Step 3. Synthesis of N-(3-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 7). Title compound was obtained using intermediate D.2 (60.0 mg, 0.14 mmol) following the general procedure 4 previously described for 4 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 60/40) afforded pure title compound (30.2 mg, yield 52%). Rt = 1.68 min (gradient 1); MS (ESI) m/z: 322.1 [M + H]+, [M + H]+ calculated for C18H17FN5: 322.1. HRMS (ESI) m/z: 322.1463 calcd for C18H17FN5 [M + H]+; found m/z: 322.1468. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (td, J = 4.2, 1.6 Hz, 2 H), 8.57 (d, J = 5.3 Hz, 1 H), 8.10 (td, J = 4.6, 1.6 Hz, 2 H), 7.74 (dt, J = 12.1, 2.3 Hz, 1 H), 7.62 (ddd, J = 8.3, 2.0, 0.9 Hz, 1 H), 7.50–7.31 (m, 1 H), 6.90 (tt, J = 8.4, 2.4 Hz, 1 H), 3.82 (d, J = 3.6 Hz, 2 H), 3.02 (td, J = 5.9, 2.6 Hz, 2 H), 2.74 (q, J = 5.4 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ 162.4 (Cq), 162.0 (Cq, d, JCF = 241.3 Hz), 157.5 (Cq), 156.62(Cq), 150.2 (CH), 145.13 (Cq), 141.50 (Cq, d, JCF = 11.1 Hz), 129.83 (CH, d, JCF = 9.5 Hz), 121.23 (CH), 117.21 (CH, d, JCF = 2.4 Hz), 114.34 (C), 109.14 (CH, d, JCF = 20.8 Hz), 108.12 (CH, d, JCF = 26.2 Hz), 42.60 (CH2), 42.25 (CH2), 32.07 (CH2). QC purity (UV at 215 nm) 99%.
Synthesis of compound 8. N-(3-fluorophenyl)-2-(4-pyridyl)pyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 2-(4-pyridyl)-3 H-pyrido[4,3-d]pyrimidin-4-one (intermediate J).
A mixture of 4-Amino-nicotinic acid (137 mg, 0.96 mmol) and pyridine-4-carboximidamide (582.8 mg, 4.81 mmol) in 1-metil-2-pirrolidone (1 ml) was stirred under Ar atmosphere at 160 °C for 72 h. The reaction crude was poured in water (10 ml), the resulting solid filtered off and the filtrate concentrated at low pressure. Final normal phase purification (DCM/DCM: MeOH 4:1 from 95/5 to 75/25) yielded pure title compound (101 mg, yield 47%). Rt = 1.10 min (gradient 1); MS (ESI) m/z: 225.4 [M + H]+, [M + H]+ calculated for C12H9N4O: 225.1. 1H NMR (400 MHz, DMSO-d6) δ 13.10 (s, 1 H), 9.33 (s, 1 H), 8.88 (d, J = 5.6 Hz, 1 H), 8.82 (d, J = 5.1 Hz, 2 H), 8.10 (d, J = 5.2 Hz, 2 H), 7.68 (d, J = 5.6 Hz, 1 H).
Step 2. Synthesis of 4-chloro-2-(4-pyridyl)pyrido[4,3-d]pyrimidine (intermediate K).
A mixture of intermediate J (85 mg, 0.38 mmol), PCl5 (88.6 mg, 0.42 mmol) and POCl3 (1.1 ml, 11.37 mmol) was stirred under N2 at 120 °C for 16 h. The reaction crude was then concentrated to dryness at low pressure, portioned between DCM (5 ml) and cold NaHCO3 saturated solution (15 ml), the organic layer dried over Na2SO4 and concentrated to dryness at low pressure yielding pure title compound (92 mg, yield 99%). Rt = 1.74 min (gradient 1); MS (ESI) m/z: 243.3/245.3 [M + H]+, [M + H]+ calculated for C12H8ClN4: 243.1/245.1. 1H NMR (400 MHz, Acetone-d6) δ 9.72 (d, J = 0.9 Hz, 1 H), 9.10 (d, J = 5.9 Hz, 1 H), 8.92–8.80 (m, 2 H), 8.49–8.40 (m, 2 H), 8.05 (dd, J = 5.9, 0.9 Hz, 1 H).
Step 3. N-(3-fluorophenyl)-2-(4-pyridyl)pyrido[4,3-d]pyrimidin-4-amine (compound 8).
Title compound was obtained using intermediate K (92 mg, 0.38 mmol) and 3-fluoroaniline (0.045 ml, 0.45 mmol) following the general procedure 2 previously described. Final normal phase purification (DCM/DCM: MeOH 4:1 from 100:0 to 60:40) yielded pure titled compound (61 mg, yield 50%). Rt = 1.93 min (gradient 1); MS (ESI) m/z: 318.4 [M + H]+, [M + H]+ calculated for C18H13FN5: 318.1. 1H NMR (400 MHz, DMSO-d6) δ 10.51 (s, 1 H), 9.92–9.80 (m, 1 H), 8.91–8.81 (m, 1 H), 8.81–8.71 (m, 2 H), 8.22 (dq, J = 4.9, 1.6 Hz, 2 H), 7.93–7.86 (m, 1 H), 7.80–7.71 (m, 1 H), 7.53 (tdd, J = 8.2, 6.7, 1.3 Hz, 1 H), 7.06 (td, J = 8.5, 2.6 Hz, 1 H). 13C NMR (101 MHz, DMSO-d6) δ 159.6 (Cq, d, JCF = 276.1 Hz), 153.71 (Cq), 151.1 (CH), 150.4 (CH), 148.3 (CH), 144.6 (Cq), 140.1 (CH, d, JCF = 10.9 Hz), 130.2 (CH, d, JCF = 9.4 Hz), 121.9 (CH), 120.7 (CH), 118.3 (CH, d, JCF = 2.6 Hz), 111.0 (CH, d, JCF = 21.2 Hz), 110.4 (Cq), 109.4 (CH, d, JCF = 25.7 Hz). QC purity (UV at 215 nm) > 99.5%.
Synthesis of compound 9. N-(3-fluorophenyl)-N-methyl-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-(4-pyridyl)-3,5,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-one (intermediate F.1).
A suspension of methyl 1-benzyl-4-oxo-3-piperidinecarboxylate hydrochloride (250 mg, 0.84 mmol), pyridine-4-carboximidamide hydrochloride (680 mg, 4.18 mmol) and sodium methoxide (251 mg, 4.60 mmol) in ethanol (3.8 ml) was stirred at reflux temperature for 16 h. Afterwards, the reaction crude was concentrated to dryness at low pressure, triturated in water (40 ml) and filtered. Resulting solid was then rinsed with methanol (0.5 ml). Final normal phase purification, (DCM/DCM: MeOH 4:1 from 90/10 to 70/30) afforded pure title compound (185 mg, yield 69%). Rt = 1.47 min (gradient 1); MS (ESI) m/z: 319.1 [M + H]+, [M + H]+ calculated for C19H18N4O: 319.1. 1H NMR (400 MHz, DMSO-d6) δ 12.82 (s, 1 H), 8.77–8.73 (m, 2 H), 8.03–7.98 (m, 2 H), 7.40–7.34 (m, 4 H), 7.32–7.24 (m, 1 H), 3.70 (s, 2 H), 3.27 (s, 2 H), 2.74 (s, 4 H).
Step 2. Synthesis of 6-benzyl-4-chloro-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidine (intermediate G.1).
Title compound was obtained using Intermediate F.1 (180.0 mg, 0.57 mmol) following the general procedure 2 previously described. Normal phase purification (DCM/DCM: MeOH 4:1 from 100/0 to 80/20) afforded pure title compound (145 mg, yield 74%). Rt = 2.63 min (gradient 1); MS (ESI) m/z: 337/339 [M + H]+, [M + H]+ calculated for C19H18ClN4: 337/339. 1H NMR (400 MHz, DMSO-d6) δ 8.79–8.73 (m, 2 H), 8.20–8.14 (m, 2 H), 7.46–7.34 (m, 4 H), 7.34 (s, 1 H), 3.81 (s, 2 H), 3.67 (s, 2 H), 3.04 (t, J = 5.8 Hz, 2 H), 2.88 (d, J = 5.9 Hz, 2 H).
Step 3. Synthesis of 6-benzyl-2-(4-pyridyl)-3,5,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-one (intermediate H.1).
A mixture of Pd(OAc)2 (9.7 mg, 0.04 mmol) and rac-BINAP (26.8 mg, 0.04 mmol) in 1,4-dioxane (1.4 ml) was stirred under Ar flushing for 10 min. Then were stepwise added a solution of corresponding intermediate G.1 (142 mg, 0.42 mmol) in 1,4-dioxane (0.7 ml), 3-fluoro-N-methylaniline (65.3 mg, 0.51 mmol) and Cs2CO3 (194.3 mg, 0.59 mmol). The reaction mixture was stirred in a CEM® microwave apparatus at 100 °C for 3 h, filtrated through a celite coarse patch, rinsed with DCM and concentrated to dryness at low pressure. Final normal phase purification (cHexane/AcOEt from 60/40 to 40/60) yielded title compound (66 mg, yield 37%). Rt = 1.71 min (gradient 2); MS (ESI) m/z: 426.5 [M + H]+, [M + H]+ calculated for C26H25FN5: 426.2. 1H NMR (400 MHz, DMSO-d6) δ 8.76–8.69 (m, 2 H), 8.28–8.22 (m, 2 H), 7.44–7.32 (m, 1 H), 7.32–7.20 (m, 3 H), 7.09 (tdd, J = 8.5, 2.5, 0.8 Hz, 1 H), 7.05–6.96 (m, 3 H), 6.89 (ddd, J = 8.0, 2.1, 0.9 Hz, 1 H), 3.49 (s, 3 H), 3.40 (s, 2 H), 2.90 (t, J = 6.0 Hz, 2 H), 2.72–2.63 (m, 2 H), 2.59 (s, 2 H).
Step 4. Synthesis of N-(3-fluorophenyl)-N-methyl-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 9).
Title compound was obtained using intermediate H.1 (63.0 mg, 0.15 mmol) following the general procedure 4 previously described for 6 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 100/0 to 85/15) afforded pure title compound (12.8 mg, yield 25%). Rt = 1.56 min (gradient 1); MS (ESI) m/z: 336.4 [M + H]+, [M + H]+ calculated for C19H19FN5: 336.2. HRMS (ESI) m/z: 336.1619 calcd for C19H19FN5 [M + H]+; found m/z: 336.1625. 1H NMR (400 MHz, DMSO- d6) δ 8.76–8.70 (m, 2 H), 8.27–8.22 (m, 2 H), 7.39 (td, J = 8.2, 6.8 Hz, 1 H), 7.12 (dt, J = 10.8, 2.3 Hz, 1 H), 7.03 (tdd, J = 8.5, 2.6, 0.8 Hz, 1 H), 6.95 (ddd, J = 8.1, 2.1, 0.9 Hz, 1 H), 3.54 (s, 3 H), 3.32 (s, 3 H), 2.95 (s, 2 H), 2.89 (t, J = 6.1 Hz, 2 H), 2.77 (t, J = 6.0 Hz, 2 H), 2.30 (s, 1 H). 13C NMR (101 MHz, DMSO-d6) δ 164.56 (Cq), 162.61 (Cq, d, JCF = 244.0 Hz), 160.60 (Cq), 158.02 (Cq), 150.27 (CH), 148.40 (Cq, d, JCF = 9.8 Hz), 144.89 (Cq), 130.84 (CH, d, JCF = 9.5 Hz), 121.42 (CH), 120.18 (CH, d, JCF = 2.6 Hz), 118.67 (Cq), 111.65 (CH, d, JCF = 20.9 Hz), 111.33 (CH, d, JCF = 23.1 Hz), 45.29 (CH2), 42.49 (CH2), 40.81 (CH3), 32.14 (CH2). QC purity (UV at 215 nm) 96%.
Synthesis of compound 10. N-(3-fluorophenyl)-2-phenyl-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-N-(3-fluorophenyl)-2-phenyl-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate D.3).
Title compound was obtained using intermediate C.2 (125 mg, 0.34 mmol) and phenyl boronic acid (51.1 mg, 0.41 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 100/0 to 80/20) afforded pure title compound (108.0 mg, yield 78%). Rt = 2.22 min (gradient 2); MS (ESI) m/z: 411.3 [M + H]+, [M + H]+ calculated for C26H24FN4: 411.2. 1H NMR (400 MHz, DMSO-d6) δ 8.58 (s, 1 H), 8.32–8.23 (m, 2 H), 7.74 (dt, J = 12.2, 2.3 Hz, 1 H), 7.57 (ddd, J = 8.2, 2.0, 0.9 Hz, 1 H), 7.51–7.45 (m, 3 H), 7.44–7.33 (m, 6 H), 7.32–7.24 (m, 1 H), 6.88 (tdd, J = 8.5, 2.6, 0.9 Hz, 1 H), 3.79 (s, 2 H), 3.64 (s, 2 H), 2.84 (t, J = 5.7 Hz, 2 H), 2.77 (t, J = 5.7 Hz, 2 H).
Step 2. Synthesis of N-(3-fluorophenyl)-2-phenyl-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 10).
Title compound was obtained using intermediate D.3 (105 mg, 0.26 mmol) following the general procedure 4 previously described for 3 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (12.8 mg, yield 25%). Rt = 1.98 min (gradient 1); MS (ESI) m/z: 321.2 [M + H]+, [M + H]+ calculated for C19H18FN4: 321.1. HRMS (ESI) m/z: 321.1510 calcd for C19H18FN4: [M + H]+; found m/z: 321.1517. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (s, 1 H), 8.34–8.22 (m, 2 H), 7.81 (dt, J = 12.2, 2.3 Hz, 1 H), 7.62 (ddd, J = 8.3, 2.1, 0.9 Hz, 1 H), 7.55–7.43 (m, 3 H), 7.39 (td, J = 8.2, 6.9 Hz, 1 H), 6.87 (td, 1 H), 3.81 (s, 2 H), 3.02 (t, J = 5.8 Hz, 2 H), 2.72 (t, J = 5.8 Hz, 2 H), 2.61 (s, 1 H). 13 C NMR (151 MHz, DMSO-d6) δ 162.28 (C), 162.04 (CF, d, JCF = 240.1 Hz), 159.33 (C), 156.49 (C), 141.86 (C, d, JCF = 11.1 Hz), 138.02 (C), 130.06 (CH), 129.83 (CH, d, JCF = 9.5 Hz), 128.45 (CH), 127.28 (CH), 116.93 (d, JCF = 1.9 Hz), 112.89 (C), 108.81 (CH, d, JCF = 21.7 Hz), 107.83 (CH, d, JCF = 26.4 Hz), 42.57 (CH2), 42.41 (CH2), 32.20 (CH2). QC purity (UV at 215 nm) 99%.
Synthesis of compound 11. N-(3-fluorophenyl)-2-(3-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-N-(3-fluorophenyl)-2-(3-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate D.4).
Title compound was obtained using intermediate C.2 (125 mg, 0.34 mmol) and pyridine-3-boronic acid (55.5 mg, 0.41 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 70/30 to 50/50) afforded pure title compound (115.8 mg, yield 83%). Rt = 1.54 min (gradient 2); MS (ESI) m/z: 412.3 [M + H]+, [M + H]+ calculated for C25H22FN5: 412.2. 1H NMR (400 MHz, DMSO-d6) δ 9.37 (dd, J = 2.2, 0.8 Hz, 1 H), 8.72–8.61 (m, 2 H), 8.51 (dt, J = 8.0, 1.9 Hz, 1 H), 7.70 (dt, J = 12.1, 2.3 Hz, 1 H), 7.55 (ddd, J = 8.3, 2.0, 0.9 Hz, 1 H), 7.51 (ddd, J = 8.0, 4.8, 0.9 Hz, 1 H), 7.46–7.33 (m, 5 H), 7.32–7.25 (m, 1 H), 6.90 (tdd, J = 8.5, 2.6, 0.9 Hz, 1 H), 3.80 (s, 2 H), 3.64 (s, 2 H), 2.86 (t, J = 5.8 Hz, 2 H), 2.78 (t, J = 5.6 Hz, 2 H).
Step 2. N-(3-fluorophenyl)-2-(3-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 11).
Title compound was obtained using intermediate D.4 (110 mg, 0.27 mmol) following the general procedure 4 previously described for 6 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (55.8 mg, yield 65%). Rt = 1.54 min (gradient 1); MS (ESI) m/z: 322.2 [M + H]+, [M + H]+ calculated for C18H17FN5: 322.1. HRMS (ESI) m/z: 322.1463 calcd for C18H17FN5 [M + H]+; found m/z: 322.1468. 1H NMR (400 MHz, DMSO-d6) δ 9.39 (dd, J = 2.2, 0.9 Hz, 1 H), 8.65 (dd, J = 4.8, 1.7 Hz, 1 H), 8.59–8.49 (m, 2 H), 7.76 (dt, J = 12.1, 2.3 Hz, 1 H), 7.61 (ddd, J = 8.3, 2.1, 0.9 Hz, 1 H), 7.51 (ddd, J = 8.0, 4.8, 0.9 Hz, 1 H), 7.40 (td, J = 8.3, 6.9 Hz, 1 H), 6.89 (tdd, J = 8.5, 2.6, 0.9 Hz, 1 H), 3.82 (s, 2 H), 3.03 (t, J = 5.8 Hz, 2 H), 2.74 (t, J = 5.8 Hz, 2 H). 13C NMR (151 MHz, DMSO-d6) δ 162.4 (Cq), 162.0 (Cq, d, JCF = 240.1 Hz), 157.7 (CH), 156.5 (CH), 150.7 (Cq), 148.6 (Cq, d, JCF = 11.1 Hz), 141.6 (CH, d, J = 11.0 Hz), 134.6 (CH), 133.3 (C), 129.9 (CH, d, J = 9.1 Hz), 123.6 (CH), 117.2 (C), 113.5 (C), 109.1 (CH, d, J = 21.4 Hz), 108.1 (CH, d, J = 26.3 Hz), 42.5 (CH2), 42.3 (CH2), 32.1 (CH2). QC purity (UV at 215 nm) 97%.
Synthesis of compound 12. N-(3-fluorophenyl)-2-(2-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-(2-pyridyl)-3,5,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-one (intermediate F.2).
A suspension of methyl 1-benzyl-4-oxo-3-piperidinecarboxylate hydrochloride (250 mg, 0.84 mmol), Pyridine-2-carboximidamideydrochloride (680 mg, 4.18 mmol) and sodium methoxide (251 mg, 4.60 mmol) in ethanol (3.8 ml) was stirred at reflux temperature for 6 h. Afterwards, the reaction crude was concentrated to dryness at low pressure, triturated in water (40 ml) and filtered. Resulting solid was then rinsed with methanol (0.5 ml). Final normal phase purification, (DCM/DCM: MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (185 mg, yield 69%). Rt = 1.81 min (gradient 1); MS (ESI) m/z: 319.1 [M + H]+, [M + H]+ calculated for C19H19N4O: 319.1. 1 H NMR (400 MHz, DMSO- d6) δ 11.95 (s, 1 H), 8.72 (dt, J = 4.8, 1.4 Hz, 1 H), 8.28 (dt, J = 7.9, 1.1 Hz, 1 H), 8.02 (td, J = 7.8, 1.8 Hz, 1 H), 7.62 (ddd, J = 7.6, 4.8, 1.2 Hz, 1 H), 7.33–7.26 (m, 1 H), 3.70 (s, 2 H), 3.28 (s, 2 H), 2.74 (s, 4 H).
Step 2. Synthesis of 6-benzyl-4-chloro-2-(2-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidine (intermediate G.2).
Title compound was obtained using Intermediate F.2 (180.0 mg, 0.57 mmol) following the general procedure 2 previously described. Normal phase purification (DCM/DCM: MeOH 4:1 from 90/10 to 0/100) afforded pure title compound (133 mg, yield 70%). Rt = 2.34 min (gradient 1); MS (ESI) m/z: 337.1/339.1 [M + H]+, [M + H]+ calculated for C19H18ClN4: 337.1/339.1. 1H NMR (400 MHz, DMSO-d6) δ 8.75 (ddd, J = 4.7, 1.8, 0.9 Hz, 1 H), 8.33 (dt, J = 8.0, 1.1 Hz, 1 H), 7.98 (td, J = 7.8, 1.8 Hz, 1 H), 7.55 (ddd, J = 7.6, 4.7, 1.2 Hz, 1 H), 7.44–7.35 (m, 4 H), 7.35–7.26 (m, 1 H), 3.81 (s, 2 H), 3.66 (s, 2 H), 3.03 (t, J = 5.8 Hz, 2 H), 2.87 (t, J = 5.8 Hz, 2 H).
Step 3. Synthesis of 6-benzyl-N-(3-fluorophenyl)-2-(2-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate H.2).
A mixture of Pd(OAc)2 (8.8 mg, 0.04 mmol) and rac-BINAP (24.5 mg, 0.04 mmol) in 1,4-dioxane (1.2 ml) was stirred under Ar flushing for 10 min. Then were stepwise added a solution of corresponding intermediate G.2 (130 mg, 0.39 mmol) in 1,4-dioxane (0.7 ml), m-fluoroaniline (0.046 ml, 0.46 mmol) and Cs2CO3 (177.8 mg, 0.54 mmol). The reaction mixture was stirred in a CEM® microwave apparatus at 100 °C for 3.5 h, filtrated through a celite coarse patch, rinsed with DCM and concentrated to dryness at low pressure. Final normal phase purification (cHexane/AcOEt from 85/15 to 0/100) yielded title compound (95 mg, yield 60%). Rt = 1.18 min (gradient 2); MS (ESI) m/z: 412.4 [M + H]+, [M + H]+ calculated for C25H23FN5: 412.2. 1H NMR (400 MHz, DMSO-d6) δ 8.71 (ddd, J = 4.8, 1.8, 0.9 Hz, 1 H), 8.61 (s, 1 H), 8.23 (dt, J = 7.9, 1.1 Hz, 1 H), 8.01 (dt, J = 12.5, 2.3 Hz, 1 H), 7.92 (td, J = 7.7, 1.8 Hz, 1 H), 7.63 (ddd, J = 8.3, 2.1, 0.9 Hz, 1 H), 7.46 (ddd, J = 7.6, 4.7, 1.2 Hz, 1 H), 7.44–7.39 (m, 2 H), 7.36 (dt, J = 9.4, 7.7 Hz, 3 H), 7.31–7.25 (m, 1 H), 6.85 (tdd, J = 8.4, 2.6, 0.9 Hz, 1 H), 3.80 (s, 2 H), 3.67 (s, 2 H), 2.85 (t, J = 5.7 Hz, 2 H), 2.78 (t, J = 5.7 Hz, 2 H).
Step 4. Synthesis of N-(3-fluorophenyl)-2-(2-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 12).
Title compound was obtained using intermediate H.2 (92.0 mg, 0.22 mmol) following the general procedure 4 previously described for 6 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 50/50 to 30/70) afforded pure title compound (34.0 mg, yield 47%). Rt = 1.55 min (gradient 1); MS (ESI) m/z: 322.4 [M + H]+, [M + H]+ calculated for C18H17FN5: 322.1. HRMS (ESI) m/z: 322.1463 calcd for C18H17FN5 [M + H]+; found m/z: 322.1475. 1H NMR (400 MHz, DMSO- d6) δ 8.72 (ddd, J = 4.7, 1.9, 0.9 Hz, 1 H), 8.59 (s, 1 H), 8.24 (dt, J = 7.9, 1.1 Hz, 1 H), 8.07 (dt, J = 12.5, 2.3 Hz, 1 H), 7.93 (td, J = 7.7, 1.8 Hz, 1 H), 7.78–7.61 (m, 1 H), 7.47 (ddd, J = 7.5, 4.7, 1.2 Hz, 1 H), 7.36 (td, J = 8.3, 7.0 Hz, 1 H), 6.85 (tdd, J = 8.4, 2.5, 0.9 Hz, 1 H), 3.93 (s, 2 H), 3.13 (t, J = 5.8 Hz, 2 H), 2.81 (t, J = 5.9 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ 162.1 (Cq, d, JCF = 240.0 Hz), 161.8 (Cq), 159.55 (Cq), 156.6 (Cq), 155.28 (C), 149.5 (CH), 141.9 (C, d, JCF = 11.3 Hz), 136.8 (CH), 129.7 (CH, d, JCF = 9.7 Hz), 124.5 (CH), 122.87 (CH), 116.7 (CH d, JCF = 12.0 Hz), 112.6 (C), 108.7 (CH, d, JCF = 20.3 Hz), 107.8 (CH, d, JCF = 26.5 Hz), 42.1 (CH2), 41.9 (CH2), 31.4 (CH2). QC purity (UV at 215 nm) 99%.
Synthesis of compound 13. N-phenyl-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-chloro-N-phenyl-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate C.3).
Title compound was obtained using intermediate B (200 mg, 0.68 mmol) and aniline (0.07 ml, 0.75 mmol) following the general procedure 2 previously described at 120 °C for 16 h. Final normal phase purification (cyclohexane/AcOEt from 95:5 to 60:40) afforded pure title compound (160 mg, yield 62%). Rt = 1.43 min (gradient 2); MS (ESI) m/z: 351.1/353.1 [M + H]+, [M + H]+ calculated for C20H20ClN4: 351.1/353.1. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (s, 1 H), 7.57–7.48 (m, 2 H), 7.43–7.32 (m, 6 H), 7.32–7.24 (m, 1 H), 7.17–7.06 (m, 1 H), 3.76 (s, 2 H), 3.53 (s, 2 H), 2.72 (s, 4 H).
Step 2. Synthesis of 6-benzyl-N-phenyl-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (D.5).
Title compound was obtained using intermediate C.3 (155.0 mg, 0.44 mmol) and Pyridine-4-boronic acid (72.4 mg, 0.53 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 50/50 to 30/70) afforded pure title compound (106.0 mg, yield 61%). Rt = 1.51 min (gradient 2); MS (ESI) m/z: 394.2 [M + H]+, [M + H]+ calculated for C25H24N5: 394.2. 1H NMR (400 MHz, DMSO-d6) δ 8.71–8.65 (m, 2 H), 8.57 (s, 1 H), 8.13–8.06 (m, 2 H), 7.74–7.65 (m, 2 H), 7.47–7.32 (m, 6 H), 7.32–7.25 (m, 1 H), 7.16–7.05 (m, 1 H), 3.80 (s, 2 H), 3.64 (s, 2 H), 2.84 (t, J = 5.5 Hz, 2 H), 2.78 (t, J = 5.4 Hz, 2 H).
Step 3. Synthesis of N-phenyl-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 13).
Title compound was obtained using intermediate D.5 (105.0 mg, 0.27 mmol) following the general procedure 4 previously described for 5 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (65.6 mg, yield 81%). Rt = 1.43 min (gradient 1); MS (ESI) m/z: 304.2 [M + H]+, [M + H]+ calculated for C18H18N5: 304.1. HRMS (ESI) m/z: 304.1557 calcd for C18H18N5 [M + H]+; found m/z: 304.1565. 1H NMR (400 MHz, DMSO-d6) δ 8.75–8.60 (m, 2 H), 8.43 (s, 1 H), 8.18–8.05 (m, 2 H), 7.80–7.71 (m, 2 H), 7.45–7.35 (m, 2 H), 7.10 (tt, J = 7.3, 1.2 Hz, 1 H), 3.81 (s, 2 H), 3.03 (t, J = 5.8 Hz, 2 H), 2.73 (t, J = 5.8 Hz, 2 H), 2.62 (s, 1 H). 13C NMR (151 MHz, DMSO-d6) δ 162.0 (Cq), 157.5 (Cq), 156.9 (Cq), 150.2 (CH), 145.3 (Cq), 139.5 (Cq), 128.4 (CH), 123.1 (CH), 122.0 (CH), 121.3 (CH), 113.9 (Cq), 42.7 (CH2), 42.3 (CH2), 32.1 (CH2). QC purity (UV at 215 nm) > 99.5%.
Synthesis of compound 14. N-(4-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-chloro-N-(4-fluorophenyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate C.4).
Title compound was obtained using intermediate B (225 mg, 0.76 mmol) and p-fluoroaniline (0.082 ml, 0.84 mmol) following the general procedure 2 previously described at 120 °C for 65 h. Final normal phase purification (cyclohexane/AcOEt from 95/5 to 75/25) afforded pure title compound (234 mg, yield 83%). Rt = 1.38 min (gradient 2); MS (ESI) m/z: 369.2/371.2 [M + H]+, [M + H]+ calculated for C20H19ClFN4: 369.1/371.1. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (s, 1 H), 7.59–7.47 (m, 2 H), 7.44–7.32 (m, 4 H), 7.32–7.24 (m, 1 H), 7.23–7.13 (m, 2 H), 3.75 (s, 2 H), 3.50 (s, 2 H), 2.71 (d, J = 2.7 Hz, 4 H).
Step 2. Synthesis of 6-benzyl-N-(4-fluorophenyl)-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate D.6).
Title compound was obtained using intermediate C.4 (230 mg, 0.62 mmol) and Pyridine-4-boronic acid (102.2 mg, 0.75 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 70/30 to 60/40) afforded pure title compound (170.0 mg, yield 70%). Rt = 1.48 min (gradient 2); MS (ESI) m/z: 412.2 [M + H]+, [M + H]+ calculated for C25H23FN5: 412.2. 1H NMR (400 MHz, DMSO- d6) δ 8.71–8.64 (m, 2 H), 8.62 (s, 1 H), 8.11–8.04 (m, 2 H), 7.76–7.62 (m, 2 H), 7.48–7.39 (m, 2 H), 7.37 (dd, J = 8.4, 6.7 Hz, 2 H), 7.33–7.26 (m, 1 H), 7.25–7.18 (m, 2 H), 3.79 (s, 2 H), 3.61 (s, 2 H), 2.84 (t, J = 5.5 Hz, 2 H), 2.78 (t, J = 5.3 Hz, 2 H).
Step 3. Synthesis of N-(4-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 14).
Title compound was obtained using intermediate D.6 (176 mg, 0.43 mmol) following the general procedure reaction 4 previously described for 5 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (72.9 mg, yield 53%). Rt = 1.51 min (gradient 1); MS (ESI) m/z: 322.1 [M + H]+, [M + H]+ calculated for C18H17FN5: 322.1. HRMS (ESI) m/z: 322.1463 calcd for C18H18N5 [M + H]+; found m/z: 322.1465. 1H NMR (400 MHz, DMSO-d6) δ 8.74–8.63 (m, 2 H), 8.50 (s, 1 H), 8.13–8.05 (m, 2 H), 7.79–7.68 (m, 2 H), 7.28–7.18 (m, 2 H), 3.79 (s, 2 H), 3.02 (t, J = 5.8 Hz, 2 H), 2.72 (t, J = 5.8 Hz, 2 H), 2.63 (s, 1 H). 13C NMR (101 MHz, DMSO-d6) δ 161.9 (Cq), 158.1 (Cq, d, JCF = 240.2 Hz), 156.9 (Cq), 150.17 (CH), 145.2 (Cq), 135.8 (Cq, d, JCF = 3.1 Hz), 124.0 (CH, d, JCF = 8.0 Hz), 121.3 (CH), 114.9 (CH, d, JCF = 22.2 Hz), 113.7 (Cq), 42.61 (CH2), 42.3 (CH2), 32.0 (CH2). QC purity (UV at 215 nm) 99%.
Synthesis of compound 15. N-(p-tolyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-chloro-N-(p-tolyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate C.5).
Title compound was obtained using intermediate B (200 mg, 0.68 mmol) and p-toluidine (81 mg, 0.75 mmol) following the general procedure 2 previously described at 100 °C for 16 h. Final normal phase purification (cyclohexane/AcOEt from 95:5 to 70:30) afforded pure title compound (235.6 mg, yield 95%). Rt = 1.55 min (gradient 2); MS (ESI) m/z: 364.1/366.1 [M + H]+, [M + H]+ calculated for C21H22ClN4+: 364.1/366.1. 1H NMR (400 MHz, CDCl3) δ 7.43–7.28 (m, 7 H), 7.14 (d, J = 8.1 Hz, 2 H), 3.82 (s, 2 H), 3.48 (s, 2 H), 2.94–2.80 (m, 4 H), 2.33 (s, 3 H).
Step 2. Synthesis of 6-benzyl-N-(p-tolyl)-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate D.7).
Title compound was obtained using intermediate C.5 (235 mg, 0.64 mmol) and Pyridine-4-boronic acid (114.7 mg, 0.77 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 80/20 to 50/50) afforded pure title compound (170 mg, yield 65%). Rt = 1.61 min (gradient 2); MS (ESI) m/z: 408.2 [M + H]+, [M + H]+ calculated for C26H26N5: 408.5. 1H NMR (400 MHz, CDCl3) δ 8.78–8.61 (m, 2 H), 8.26–8.11 (m, 2 H), 7.54–7.46 (m, 2 H), 7.45–7.34 (m, 4 H), 7.34–7.28 (m, 1 H), 7.20 (d, J = 8.2 Hz, 2 H), 6.08 (s, 1 H), 3.82 (s, 2 H), 3.51 (s, 2 H), 3.00 (t, J = 5.8 Hz, 2 H), 2.90 (t, J = 5.7 Hz, 2 H), 2.37 (s, 3 H).
Step 3. Synthesis of N-(p-tolyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 15).
Title compound was obtained using intermediate D.7 (170 mg, 0.42 mmol) following the general procedure 4 previously described for 5 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 20/80) afforded pure title compound (29.7 mg, yield 22%). Rt = 1.57 min (gradient 1); MS (ESI) m/z: 318.2 [M + H]+, [M + H]+ calculated for C19H20N5: 318.2. HRMS (ESI) m/z: 318.1713 calcd for C19H20N5 [M + H]+; found m/z: 318.1725. 1H NMR (400 MHz, DMSO-d6) δ 8.72–8.63 (m, 2 H), 8.42 (s, 1 H), 8.12–8.07 (m, 2 H), 7.64–7.58 (m, 2 H), 7.20 (d, J = 8.1 Hz, 2 H), 3.82 (s, 2 H), 3.05 (t, J = 5.8 Hz, 2 H), 2.74 (t, J = 5.7 Hz, 2 H), 2.31 (s, 3 H). 13C NMR (101 MHz, DMSO-d6) δ 161.51 (Cq), 157.6 (Cq), 157.0 (Cq), 150.2 (CH), 145.3 (Cq), 136.8 (Cq), 132.10 (Cq), 128.80 (CH), 122.16 (CH), 121.29 (CH), 113.15 (Cq), 42.36 (CH2), 42.1 (CH2), 31.7 (CH2), 20.5 (CH3). QC purity (UV at 215 nm) > 99.5%.
Synthesis of compound 16. N-(m-tolyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-chloro-N-(m-tolyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate C.6).
Title compound was obtained using intermediate B (200 mg, 0.68 mmol) and m-toluidine (0.083 ml, 0.75 mmol) following the general procedure 2 previously described at 100 °C for 4 h. Final normal phase purification (cyclohexane/TBME from 100/0 to 70/30) afforded pure title compound (129 mg, yield 52%). Rt = 1.64 min (gradient 2); MS (ESI) m/z: 365.2/367.2 [M + H]+, [M + H]+ calculated for C21H22ClN4: 365.9/367.9.
Step 2. Synthesis of 6-benzyl-N-(m-tolyl)-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate D.8).
Title compound was obtained using intermediate C.6 (126 mg, 0.35 mmol) and pyridine-4-boronic acid (56.6 mg, 0.41 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 60/40 to 20/80) afforded pure title compound (66.0 mg, yield 47%). Rt = 0.79 min (gradient 2); MS (ESI) m/z: 408.2 [M + H]+, [M + H]+ calculated for C26H26N5: 408.2. 1H NMR (400 MHz, DMSO-d6) δ 8.73–8.65 (m, 2 H), 8.50 (s, 1 H), 8.15–8.04 (m, 2 H), 7.56–7.50 (m, 2 H), 7.45–7.39 (m, 2 H), 7.37 (dd, J = 8.4, 6.7 Hz, 2 H), 7.32–7.23 (m, 2 H), 6.96–6.89 (m, 1 H), 3.79 (s, 2 H), 3.62 (s, 2 H), 2.84 (t, J = 5.6 Hz, 2 H), 2.78 (t, J = 5.3 Hz, 2 H), 2.34 (s, 3 H).
Step 3. Synthesis of N-(m-tolyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 16).
Title compound was obtained using intermediate D.9 (58 mg, 0.14 mmol) following the general procedure 4 previously described for 6 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 55/45) afforded pure title compound (15.4 mg, yield 34%). Rt = 1.56 min (gradient 1); MS (ESI) m/z: 318.2 [M + H]+, [M + H]+ calculated for C19H20N5: 318.2. HRMS (ESI) m/z: 318.1713 calcd for C19H20N5 [M + H]+; found m/z: 318.1718. 1H NMR (600 MHz, DMSO-d6) δ 8.70–8.65 (m, 2 H), 8.38 (s, 1 H), 8.18–8.05 (m, 2 H), 7.66–7.52 (m, 2 H), 7.27 (t, J = 7.7 Hz, 1 H), 6.92 (dq, J = 7.6, 0.9 Hz, 1 H), 3.80 (d, J = 1.4 Hz, 2 H), 3.32 (s, 2 H), 3.02 (t, J = 5.8 Hz, 2 H), 2.72 (s, 2 H), 2.35 (s, 3 H). 13C NMR (151 MHz, DMSO-d6) δ 161.9 (Cq), 157.4 (Cq), 156.9 (Cq), 150.2 (CH), 145.3 (Cq), 139.4 (Cq), 137.4 (Cq), 128.2 (CH), 123.7 (CH), 122.5 (CH), 121.3 (CH), 119.1 (CH), 113.9 (Cq), 42.6 (CH2), 42.3 (CH2), 32.1 (CH2), 21.2 (CH3). QC purity (UV at 215 nm) 95%.
Synthesis of compound 17. 4-[[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]amino]phenol
Step 1. Synthesis of 4-[(6-benzyl-2-chloro-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl)amino]phenol (intermediate C.7).
Title compound was obtained using intermediate B (200 mg, 0.68 mmol) and p-aminophenol (82.4 mg, 0.75 mmol) following general procedure 2 previously described at 100 °C for 16 h. Final normal phase purification (cyclohexane/AcOEt from 85/15 to 50/50) afforded pure title compound (149 mg, yield 60%). Rt = 0.74 min (gradient 2); MS (ESI) m/z: 366.1/368.1 [M + H]+, [M + H]+ calculated for C20H20ClN4O: 366.1/368.1. 1H NMR (400 MHz DMSO-d6) δ 9.32 (s, 1 H), 8.57 (s, 1 H), 7.46–7.31 (m, 4 H), 7.30–7.24 (m, 1 H), 7.23–7.12 (m, 2 H), 6.81–6.65 (m, 2 H), 3.74 (s, 2 H), 3.45 (s, 2 H), 2.78–2.59 (m, 4 H).
Step 2. 4-[[6-benzyl-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl]amino]phenol (intermediate D.9).
Title compound was obtained using intermediate C.7 (148 mg, 0.40 mmol) and Pyridine-4-boronic acid (72.1 mg, 0.49 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 60/40 to 30/70) afforded pure title compound (75 mg, yield 46%). Rt = 0.77 min (gradient 2); MS (ESI) m/z: 410.2 [M + H]+, [M + H]+ calculated for C25H24N5O: 410.2. 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1 H), 8.69–8.61 (m, 2 H), 8.37 (s, 1 H), 8.09–8.02 (m, 2 H), 7.44–7.32 (m, 6 H), 7.31–7.25 (m, 1 H), 6.78 (d, J = 8.8 Hz, 2 H), 3.77 (s, 2 H), 3.56 (s, 2 H), 2.84–2.71 (m, 4 H).
Step 3. 4-[[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]amino]phenol (compound 17).
Title compound was obtained using intermediate D.9 (75.0 mg, 0.18 mmol) following the general procedure 4 previously described for 5 h. Final normal phase purification (DCM/DCM: NH3 1 M MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (26.9 mg, yield 47%). Rt = 1.32 min (gradient 1); MS (ESI) m/z: 320.1 [M + H]+, [M + H]+ calculated for C18H18N5O: 320.1. HRMS (ESI) m/z: 320.1506 calcd for C18H18N5O [M + H]+; found m/z: 320.1508. 1H NMR (400 MHz, DMSO- d6) δ 8.71–8.61 (m, 2 H), 8.31 (s, 1 H), 8.10–8.00 (m, 2 H), 7.56–7.33 (m, 2 H), 6.89–6.69 (m, 2 H), 3.77 (s, 2 H), 3.03 (t, J = 5.8 Hz, 2 H), 2.71 (t, J = 5.8 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ 161.1 (Cq), 157.5 (Cq), 157.2 (Cq), 153.7 (Cq), 150.1 (CH), 145.4 (Cq), 130.6 (Cq), 124.4 (CH), 121.3 (CH), 114.9 (CH), 112.73 (C), 42.4 (CH2), 42.2 (CH2), 31.75 (CH2). QC purity (UV at 215 nm) 96%.
Synthesis of compound 18. 3-[[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]amino]phenol
Step 1. Synthesis of 3-[(6-benzyl-2-chloro-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl)amino]phenol (intermediate C.8).
Title compound was obtained using intermediate B (200 mg, 0.68 mmol) and 3-aminophenol (83.3 mg, 0.75 mmol) following general procedure 2 previously described at 120 °C for 24 h. Final normal phase purification (cyclohexane/AcOEt from 80/20 to 60/40) afforded pure title compound (107 mg, yield 43%). Rt = 0.82 min (gradient 2); MS (ESI) m/z: 366.1/368.1 [M + H]+, [M + H]+ calculated for C20H20ClN4O: 366.1/368.1. 1H NMR (400 MHz DMSO-d6) δ 9.41 (s, 1 H), 8.60 (s, 1 H), 7.42–7.32 (m, 4 H), 7.31–7.22 (m, 1 H), 7.11 (t, J = 8.0 Hz, 1 H), 7.00 (t, J = 2.2 Hz, 1 H), 6.95 (ddd, J = 8.0, 2.1, 1.0 Hz, 1 H), 6.52 (ddd, J = 8.1, 2.4, 1.0 Hz, 1 H), 3.75 (s, 2 H), 3.50 (s, 2 H), 2.70 (s, 4 H).
Step 2. Synthesis of 3-[[6-benzyl-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl]amino]phenol (intermediate D.10).
Title compound was obtained using intermediate C.8 (100 mg, 0.27 mmol) and Pyridine-4-boronic acid (44.7 mg, 0.33 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 70:30 to 10:90) afforded pure title compound (90 mg, yield 81%). Rt = 0.86 min (gradient 2); MS (ESI) m/z: 410.2 [M + H]+, [M + H]+ calculated for C25H24N5O: 410.2. 1H NMR (400 MHz, DMSO-d6) δ 9.38 (s, 1 H), 8.73–8.63 (m, 2 H), 8.41 (s, 1 H), 8.17–8.08 (m, 2 H), 7.45–7.39 (m, 2 H), 7.36 (dd, J = 8.4, 6.7 Hz, 2 H), 7.31–7.25 (m, 1 H), 7.22 (t, J = 2.0 Hz, 1 H), 7.19–7.12 (m, 2 H), 6.51 (dt, J = 7.0, 2.3 Hz, 1 H), 3.79 (s, 2 H), 3.62 (s, 2 H), 2.84 (d, J = 5.6 Hz, 2 H), 2.78 (d, J = 5.4 Hz, 2 H).
Step 3. Synthesis of 3-[[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]amino]phenol (compound 18).
Title compound was obtained using intermediate D.10 (85 mg, 0.21 mmol) following the general procedure 4 previously described for 5 h. Final normal phase purification (DCM/DCM: NH3 1 M MeOH 4:1 from 95/5 to 40/60) afforded pure title compound (42.4 mg, yield 64%). Rt = 1.20 min (gradient 1); MS (ESI) m/z: 320.1 [M + H]+, [M + H]+ calculated for C18H18N5O: 320.1. HRMS (ESI) m/z: 320.1506 calcd for C18H18N5O [M + H]+; found m/z: 320.1509. 1H NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1 H), 8.80–8.62 (m, 2 H), 8.33 (s, 1 H), 8.19–8.08 (m, 2 H), 7.28 (t, J = 2.1 Hz, 1 H), 7.22–7.09 (m, 2 H), 6.51 (dt, J = 7.2, 2.0 Hz, 1 H), 3.82 (s, 2 H), 3.05 (t, J = 5.8 Hz, 2 H), 2.74 (t, J = 5.8 Hz, 2 H). 13C NMR (101 MHz, DMSO-d6) δ 161.7 (Cq), 157.6 (Cq), 157.4 (Cq), 156.9 (Cq), 150.2 (CH), 145.3 (Cq), 140.5 (Cq), 129.0 (CH), 121.4 (CH), 113.4 (Cq), 112.7 (CH), 110.3 (CH), 109.0 (CH), 42.4 (CH2), 42.1 (CH2), 31.7 (CH2). QC purity (UV at 215 nm) 97%.
Synthesis of compound 19. N-(4-methoxyphenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-chloro-N-(4-methoxyphenyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate C.9).
Title compound was obtained using intermediate B (200 mg, 0.68 mmol) and p-methoxyaniline (93.0 mg, 0.75 mmol) following general procedure 2 previously described at 120 °C for 15 h. Final normal phase purification (cyclohexane/AcOEt from 90/10 to 70/30) afforded pure title compound (204 mg, yield 79%). Rt = 1.24 min (gradient 2); MS (ESI) m/z: 380.1/382.1 [M + H]+, [M + H]+ calculated for C21H22ClN4O: 380.1/382.1. 1H NMR (400 MHz DMSO-d6) δ 8.67 (s, 1 H), 7.46–7.32 (m, 6 H), 7.32–7.23 (m, 1 H), 6.98–6.87 (m, 2 H), 3.75 (bs, 5 H), 3.47 (s, 2 H), 2.74–2.65 (m, 4 H).
Step 2. Synthesis of 6-benzyl-N-(4-methoxyphenyl)-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate D.11).
Title compound was obtained using intermediate C.9 (200 mg, 0.53 mmol) and Pyridine-4-boronic acid (86.1 mg, 0.63 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 60/40 to 10/90) afforded pure title compound (171 mg, yield 77%). Rt = 1.32 min (gradient 2); MS (ESI) m/z: 424.3 [M + H]+, [M + H] + calculated for C26H26N5O+: 424.2. 1 H NMR (400 MHz, DMSO- d6) δ 8.73–8.62 (m, 2 H), 8.47 (s, 1 H), 8.15–8.04 (m, 2 H), 7.63–7.50 (m, 2 H), 7.47–7.39 (m, 2 H), 7.37 (dd, J = 8.4, 6.7 Hz, 2 H), 7.31–7.23 (m, 1 H), 7.03–6.91 (m, 2 H), 3.78 (s, 2 H), 3.77 (s, 3 H), 3.59 (s, 2 H), 2.83 (t, J = 5.7 Hz, 2 H), 2.77 (t, J = 5.4 Hz, 2 H).
Step 3. Synthesis of N-(4-methoxyphenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 19).
Title compound was obtained using intermediate D.11 (166 mg, 0.39 mmol) following the general procedure 4 previously described for 6 h. Final normal phase purification (DCM/DCM: NH3 1 M MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (73.2 mg, yield 56%). Rt = 1.46 min (gradient 1); MS (ESI) m/z: 334.2 [M + H]+, [M + H]+ calculated for C19H20N5O: 334.2. HRMS (ESI) m/z: 334.1662 calcd for C19H20N5O [M + H]+; found m/z: 334.1666. 1H NMR (400 MHz, DMSO-d6) δ 8.84–8.59 (m, 2 H), 8.36 (s, 1 H), 8.18–8.04 (m, 2 H), 7.69–7.56 (m, 2 H), 7.09–6.93 (m, 2 H), 3.79 (s, 3 H), 3.78 (s, 2 H), 3.03 (t, J = 5.7 Hz, 2 H), 2.71 (t, J = 5.8 Hz, 2 H), 2.62 (s, 1 H). 13C NMR (101 MHz, DMSO-d6) δ 161.4 (Cq), 157.5 (Cq), 157.1 (Cq), 155.4 (Cq), 150.1 (CH), 145.4 (Cq), 132.3 (Cq), 123.9 (CH), 121.3 (CH), 113.6 (CH), 113.4 (Cq), 55.2 (CH3), 42.6 (CH2), 42.4 (CH2), 32.0 (CH2). QC purity (UV at 215 nm) > 99.5%.
Synthesis of compound 20. N-(3-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-chloro-N-(3-methoxyphenyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate C.10).
Title compound was obtained using compound B (150 mg, 0.51 mmol) and m-methoxyaniline (0.07 ml, 0.56 mmol) following the general procedure 2 previously described at 100 °C for 16 h. Final normal phase purification (cyclohexane/AcOEt from 95/5 to 60/40) afforded pure title compound (150 mg, yield 58%). Rt = 1.39 min (gradient 2); MS (ESI) m/z: 381.2/383.2 [M + H]+, [M + H]+ calculated for C21H22ClN4O: 381.1/383.1. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1 H), 7.43–7.32 (m, 4 H), 7.30–7.25 (m, 1 H), 7.24 (s, 1 H), 7.24–7.20 (m, 1 H), 7.15 (ddd, J = 8.0, 2.0, 1.0 Hz, 1 H), 6.69 (ddd, J = 8.2, 2.6, 1.0 Hz, 1 H), 3.76 (s, 2 H), 3.74 (s, 3 H), 3.52 (s, 2 H), 2.71 (s, 4 H).
Step 2. Synthesis of 6-benzyl-N-(3-methoxyphenyl)-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate D.12).
Title compound was obtained using compound C.10 (145 mg, 0.38 mmol) and Pyridine-4-boronic acid (62.4 mg, 0.46 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 50/50 to 30/70) afforded pure title compound (130 mg, yield 81%). Rt = 1.52 min (gradient 2); MS (ESI) m/z: 424.3 [M + H]+, [M + H]+ calculated for C26H26N5O: 424.2. 1H NMR (400 MHz, DMSO-d6) δ 8.74–8.64 (m, 2 H), 8.52 (s, 1 H), 8.28–7.98 (m, 2 H), 7.45–7.25 (m, 8 H), 6.67 (ddd, J = 7.9, 2.5, 1.2 Hz, 1 H), 3.79 (s, 2 H), 3.78 (s, 3 H), 3.64 (s, 2 H), 2.85 (t, J = 5.8 Hz, 2 H), 2.78 (t, J = 5.6 Hz, 2 H).
Step 3. N-(3-methoxyphenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 20).
Title compound was obtained using intermediate D.12 (125.0 mg, 0.30 mmol) following the general procedure 4 previously described for 4 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (71.8 mg, yield 73%). Rt = 1.47 min (gradient 1); MS (ESI) m/z: 334.2 [M + H]+, [M + H]+ calculated for C19H20N5O: 334.2. HRMS (ESI) m/z: 334.1662 calcd for C19H20N5O [M + H]+; found m/z: 334.1666. 1H NMR (400 MHz, DMSO-d6) δ 8.74–8.66 (m, 2 H), 8.39 (s, 1 H), 8.17–8.09 (m, 2 H), 7.47 (t, J = 2.2 Hz, 1 H), 7.39 (ddd, J = 8.1, 2.0, 1.0 Hz, 1 H), 7.28 (t, J = 8.1 Hz, 1 H), 6.67 (ddd, J = 8.2, 2.6, 0.9 Hz, 1 H), 3.82 (s, 2 H), 3.78 (s, 3 H), 3.31 (s, 1 H), 3.03 (t, J = 5.8 Hz, 2 H), 2.73 (t, J = 5.8 Hz, 2 H). 13C NMR (151 MHz, DMSO-d6) δ 162.1 (Cq), 159.3 (Cq), 157.5 (Cq), 156.8 (Cq), 150.2 (CH), 145.3 (Cq), 140.8 (Cq), 129.10 (CH), 121.3 (CH), 114.1 (Cq), 113.9 (CH), 108.8 (CH), 107.1 (CH), 55.0 (CH3), 42.6 (CH2), 42.3 (CH2), 32.1 (CH2). QC purity (UV at 215 nm) 96%.
Synthesis of compound 21. N1-methyl-N4-[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]benzene-1,4-diamine
Step 1. Synthesis of tert-butyl N-[4-[(6-benzyl-2-chloro-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl)amino]phenyl]-N-methyl-carbamate (intermediate C.11).
Title compound was obtained using intermediate B (235 mg, 0.79 mmol) and 4-(N-tert-Butoxycarbonyl-N-methylamino)aniline (193 mg, 0.87 mmol) following the general procedure 2 previously described at 120 °C for 17 h. Final normal phase purification (cyclohexane/AcOEt from 70/30 to 50/50) afforded pure title compound (243 mg, yield 64%). Rt = 1.82 min (gradient 2); MS (ESI) m/z: x [M + H]+, [M + H]+ calculated for C26H31ClN5O2: 480.0/482.0.
Step 2. Synthesis of tert-butyl N-[4-[[6-benzyl-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl]amino]phenyl]-N-methyl-carbamate (intermediate D.13).
Title compound was obtained using intermediate C.11 (215 mg, 0.45 mmol) and Pyridine-4-boronic acid (73.4 mg, 0.54 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 85/15 to 50/50) afforded pure title compound (178 mg, yield 76%). Rt = 1.82 min (gradient 2); MS (ESI) m/z: 523.3 [M + H]+, [M + H]+ calculated for C31H35N6O2: 523.3. 1H NMR (400 MHz, DMSO-d6) δ 8.73–8.61 (m, 2 H), 8.57 (s, 1 H), 8.14–8.06 (m, 2 H), 7.71–7.61 (m, 2 H), 7.45–7.39 (m, 2 H), 7.39–7.32 (m, 2 H), 7.32–7.23 (m, 3 H), 3.79 (s, 2 H), 3.63 (s, 2 H), 3.20 (s, 3 H), 2.84 (d, J = 5.5 Hz, 2 H), 2.78 (t, J = 5.6 Hz, 2 H), 1.42 (s, 9 H).
Step 3. Synthesis of N1-methyl-N4-[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]benzene-1,4-diamine (compound 21).
Title compound was obtained solving intermediate D.13 (175 mg, 0.33 mmol) in 1,4-dioxane (0.8 ml), a solution of HCl (4 M) in 1,4-dioxane (0.8 ml) was dropwise added and the reaction mixture stirred at room temperature for 1 h, then the reaction crude was concentrated to dryness at low pressure, the resulting crude portioned between AcOEt (20 ml) and NaOH 0.1 M (20 ml), the organic layer dried over Na2SO4 and concentrated to dryness at low pressure. Resulting crude solid (140 mg, 0.33 mmol) was treated following the general procedure 4 previously described for 6 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (70.2 mg, yield 64%). Rt = 1.24 min (gradient 1); MS (ESI) m/z: 333.2 [M + H]+, [M + H]+ calculated for C19H21N6: 333.4. HRMS (ESI) m/z: 333.1822 calcd for C19H21N6 [M + H]+; found m/z: 333.1823. 1H NMR (400 MHz, DMSO-d6) δ 8.69–8.57 (m, 2 H), 8.15 (s, 1 H), 8.08–8.02 (m, 2 H), 7.45–7.32 (m, 2 H), 6.61–6.53 (m, 2 H), 5.47 (q, J = 5.2 Hz, 1 H), 3.73 (s, 2 H), 3.00 (t, J = 5.8 Hz, 2 H), 2.69 (m, 5 H). 13C NMR (101 MHz, DMSO-d6) δ 160.9 (Cq), 157.5 (Cq), 157.3 (Cq), 150.0 (CH), 146.6 (Cq), 145.6 (Cq), 128.0 (Cq), 124.3 (CH), 121.3 (CH), 112.9 (Cq), 111.3 (CH), 42.6 (CH2), 42.4 (CH2), 32.0 (CH2), 30.1 (CH3). QC purity (UV at 215 nm) 99%.
Synthesis of compound 22. N3-methyl-N1-[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]benzene-1,3-diamine
Step 1. Synthesis of tert-butyl N-[3-[(6-benzyl-2-chloro-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl)amino]phenyl]-N-methyl-carbamate (intermediate C.12).
Title compound was obtained using intermediate B (200 mg, 0.68 mmol) and 3-(N-tert-Butoxycarbonyl-N-methylamino)aniline (175.0 mg, 0.75 mmol) following the general procedure 2 previously described at 100 °C for 16 h. Final normal phase purification (cyclohexane/AcOEt from 85:15 to 65:35) afforded titled compound. Rt = 1.80 min (gradient 2); MS (ESI) m/z: 480.0/482.0 [M + H]+, [M + H] + calculated for C26H31ClN5O2+: 480.2/482.0. 1 H NMR (400 MHz, DMSO- d6) δ 8.74 (s, 1 H), 7.50 (t, J = 2.1 Hz, 1 H), 7.43–7.24 (m, 8 H), 7.02 (ddd, J = 7.9, 2.2, 1.0 Hz, 1 H), 3.76 (s, 2 H), 3.53 (s, 2 H), 3.18 (s, 3 H), 2.72 (s, 4 H), 1.40 (s, 9 H).
Step 2. Synthesis of tert-butyl N-[3-[[6-benzyl-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl]amino]phenyl]-N-methyl-carbamate (intermediate D.14).
Title compound was obtained using intermediate C.12 (92 mg, 0.19 mmol) and Pyridine-4-boronic acid (31.4 mg, 0.23 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 85/15 to 30/70) afforded pure title compound (86 mg, yield 86%). Rt = 1.87 min (gradient 2); MS (ESI) m/z: 523.3 [M + H]+, [M + H]+ calculated for C31H35N6O2: 523.3. 1H NMR (400 MHz, DMSO-d6) δ 8.70–8.64 (m, 2 H), 8.60 (s, 1 H), 8.16–8.10 (m, 2 H), 7.65 (t, J = 2.1 Hz, 1 H), 7.60–7.54 (m, 1 H), 7.46–7.40 (m, 2 H), 7.40–7.32 (m, 3 H), 7.32–7.25 (m, 1 H), 7.00 (ddd, J = 8.0, 2.2, 1.0 Hz, 1 H), 3.80 (s, 2 H), 3.64 (s, 2 H), 3.22 (s, 3 H), 2.85 (d, J = 5.6 Hz, 2 H), 2.78 (t, J = 5.5 Hz, 2 H), 1.40 (d, J = 1.1 Hz, 9 H).
Step 3. N3-methyl-N1-[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]benzene-1,3-diamine (compound 22).
Title compound was obtained solving intermediate D.14 (95.0 mg, 0.19 mmol) in 1,4-dioxane (0.4 ml), a solution of HCl (4 M) in 1,4-dioxane (0.4 ml) was dropwise added and the reaction mixture stirred at room temperature for 3 h, then the reaction crude was concentrated to dryness at low pressure, the resulting crude portioned between DCM (20 ml) and NaOH 0.1 M (20 ml), the organic layer dried over Na2SO4 and concentrated to dryness at low pressure. Resulting crude (66 mg, 0.16 mmol) was treated following the general procedure 4 previously described for 4 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (16.1 mg, yield 31%). Rt = 1.36 min (gradient 1); MS (ESI) m/z: 333.3 [M + H]+, [M + H]+ calculated for C19H21N6: 333.2. HRMS (ESI) m/z: 333.1822 calcd for C19H21N6 [M + H]+; found m/z: 333.1827. 1H NMR (400 MHz, DMSO-d6) δ 8.81–8.56 (m, 2 H), 8.18 (s, 1 H), 8.16–8.11 (m, 2 H), 7.11–7.00 (m, 2 H), 6.95 (ddd, J = 8.0, 2.1, 1.0 Hz, 1 H), 6.30 (ddd, J = 8.1, 2.3, 1.0 Hz, 1 H), 5.66 (q, J = 5.0 Hz, 1 H), 3.79 (s, 2 H), 3.02 (t, J = 5.8 Hz, 2 H), 2.71 (t, J = 5.6 Hz, 5 H). 13C NMR (101 MHz, DMSO-d6) δ 161.7 (Cq), 157.6 (Cq), 157.1 (Cq), 150.2 (CH), 145.5 (Cq), 140.4 (Cq), 128.7 (CH), 121.5 (CH), 109.6 (CH), 107.6 (CH), 104.9 (CH), 99.6 (C), 42.7 (CH2), 42.4 (CH2), 32.1 (CH2), 30.0 (CH3). QC purity (UV at 215 nm) 99%.
Synthesis of compound 23. N3,N3-dimethyl-N1-[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]benzene-1,3-diamine
Step 1. Synthesis of N1-(6-benzyl-2-chloro-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl)-N3,N3-dimethyl-benzene-1,3-diamine (intermediate C.13).
Title compound was obtained using intermediate B (250 mg, 0.85 mmol) and N,N-Dimethyl-1,3-phenylenediamine dihydrochloride (197.4 mg, 0.93 mmol) following the general procedure 2 previously described at 100 °C for 16 h. Final normal phase purification (cyclohexane/AcOEt from 80/20 to 60/40) afforded pure title compound (227.6 mg, yield 68%). Rt = 1.12 min (gradient 2); MS (ESI) m/z: 394.2/396.2 [M + H]+, [M + H]+ calculated for C22H24ClN5: 394.2/396.2. 1H NMR (400 MHz, DMSO-d6) δ 8.58 (s, 1 H), 7.43–7.31 (m, 4 H), 7.31–7.24 (m, 1 H), 7.13 (t, J = 8.1 Hz, 1 H), 6.95 (t, J = 2.2 Hz, 1 H), 6.88 (ddd, J = 8.0, 2.0, 0.8 Hz, 1 H), 6.50 (ddd, J = 8.4, 2.5, 0.9 Hz, 1 H), 3.75 (s, 2 H), 3.51 (s, 2 H), 2.88 (s, 6 H), 2.70 (d, J = 2.4 Hz, 4 H).
Step 2. Synthesis of N1-[6-benzyl-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-yl]-N3,N3-dimethyl-benzene-1,3-diamine (intermediate D.15).
Title compound was obtained using compound C.13 (220 mg, 0.56 mmol) and Pyridine-4-boronic acid (91.5 mg, 0.67 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 70/30 to 20/80) afforded pure title compound (185 mg, yield 76%). Rt = 1.43 min (gradient 2); MS (ESI) m/z: 437.2 [M + H]+, [M + H]+ calculated for C27H28N6: 437.2. 1H NMR (400 MHz, DMSO- d6): δ 8.70–8.64 (m, 2 H), 8.36 (s, 1 H), 8.17–8.09 (m, 2 H), 7.48–7.39 (m, 2 H), 7.39–7.33 (m, 2 H), 7.31–7.25 (m, 1 H), 7.23–7.13 (m, 2 H), 7.06 (ddd, J = 7.9, 2.0, 0.9 Hz, 1 H), 6.49 (ddd, J = 8.3, 2.6, 0.9 Hz, 1 H), 3.79 (s, 2 H), 3.63 (s, 2 H), 2.92 (s, 6 H), 2.84 (t, J = 5.2 Hz, 2 H), 2.77 (t, J = 5.4 Hz, 2 H).
Step 3. N3,N3-dimethyl-N1-[2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-yl]benzene-1,3-diamine (compound 23).
Title compound was obtained using intermediate D.15 (180.0 mg, 0.41 mmol) following the general procedure 4 previously described for 4 h. Final normal phase purification (DCM/DCM: NH3 1 N MeOH 4:1 from 70/30 to 20/80) afforded pure title compound (62.8 mg, yield 44%). Rt = 1.55 min (gradient 1); MS (ESI) m/z: 347.2 [M + H]+, [M + H]+ calculated for C20H23N6: 347.2. HRMS (ESI) m/z: 347.1979 calcd for C20H23N6 [M + H]+; found m/z: 347.1978. 1H NMR (400 MHz, DMSO-d6): δ 8.71–8.65 (m, 2 H), 8.22 (s, 1 H), 8.17–8.10 (m, 2 H), 7.25–7.13 (m, 2 H), 7.12 (dt, J = 8.2, 1.3 Hz, 1 H), 6.48 (ddd, J = 8.1, 2.5, 1.1 Hz, 1 H), 3.80 (s, 2 H), 3.02 (t, J = 5.8 Hz, 2 H), 2.92 (s, 6 H), 2.72 (t, J = 5.8 Hz, 2 H). 13C NMR (151 MHz, DMSO-d6) δ 161.8 (Cq), 157.4 (Cq), 157.0 (Cq), 150.7 (CH), 150.1 (Cq), 145.4 (Cq), 140.2 (Cq), 128.7 (CH), 121.3 (CH), 113.9 (Cq), 110.2 (CH), 107.7 (CH), 106.0 (CH), 42.7 (CH2), 42.3 (CH2), 40.3 (CH3), 32.1 (CH2). QC purity (UV at 215 nm) > 99.5%.
Synthesis of compound 24. N-(2-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine
Step 1. Synthesis of 6-benzyl-2-chloro-N-(2-fluorophenyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate C.14).
A mixture of Pd(OAc)2 (7.8 mg, 0.03 mmol) and rac-BINAP (21.6 mg, 0.03 mmol) in 1,4-dioxane (3.4 ml) was stirred under Ar flushing for 10 min. Then were stepwise added intermediate B (200 mg, 0.68 mmol), o-fluoroaniline (0.066 ml, 0.68 mmol) and Cs2CO3 (268.5 mmol, 0.82 mmol). The reaction mixture was stirred in a CEM® microwave apparatus at 80 °C for 1.5 h, filtrated through a celite coarse patch, rinsed with DCM and concentrated to dryness at low pressure. Final normal phase purification (cyclohexane/AcOEt from 95/5 to 75/25) yielded a mixture where titled compound was the majority one. Resulting crude (205 mg) was used in the next step without any further purification process. Rt = 1.25 min (gradient 2, 77% purity); MS (ESI) m/z: 367.1/369.1 [M + H]+, [M + H]+ calculated for C20H19ClFN4: 367.1/369.1.
Step 2. 6-benzyl-N-(2-fluorophenyl)-2-(4-pyridyl)-7,8-dihydro-5 H-pyrido[4,3-d]pyrimidin-4-amine (intermediate D.16).
Title compound was obtained using crude C.14 (205 mg, 0.56 mmol estimated) and Pyridine-4-boronic acid (91.1 mg, 0.67 mmol) following the general procedure 3 previously described. Final normal phase purification (cyclohexane/AcOEt from 85/15 to 45/55) afforded pure title compound (149 mg, average yield 65% from previous step). Rt = 1.37 min (gradient 2); MS (ESI) m/z: 412.2 [M + H]+, [M + H]+ calculated for C25H23FN5: 412.2. 1H NMR (400 MHz, DMSO-d6) δ 8.67–8.55 (m, 3 H), 8.00–7.88 (m, 2 H), 7.50 (td, J = 7.8, 1.8 Hz, 1 H), 7.45–7.40 (m, 2 H), 7.40–7.34 (m, 2 H), 7.34–7.20 (m, 4 H), 3.79 (s, 2 H), 3.60 (s, 2 H), 2.86 (t, J = 5.7 Hz, 2 H), 2.81 (d, J = 5.0 Hz, 2 H).
Step 3. N-(2-fluorophenyl)-2-(4-pyridyl)-5,6,7,8-tetrahydropyrido[4,3-d]pyrimidin-4-amine (compound 24).
Title compound was obtained using intermediate D.16 (140.0 mg, 0.34 mmol) following the general procedure 4 previously described for 6 h. Final normal phase purification (DCM/DCM: NH3 1 M MeOH 4:1 from 95/5 to 75/25) afforded pure title compound (72.2 mg, yield 66%). Rt = 0.33 min (gradient 2); MS (ESI) m/z: 322.2 [M + H]+, [M + H]+ calculated for C18H17FN5: 322.1. HRMS (ESI) m/z: 322.1463 calcd for C18H17FN5 [M + H]+; found m/z: 322.1476. 1H NMR (400 MHz, DMSO-d6) δ 8.67–8.58 (m, 2 H), 8.53 (s, 1 H), 8.00–7.90 (m, 2 H), 7.54 (td, J = 7.8, 2.0 Hz, 1 H), 7.38–7.20 (m, 3 H), 3.85 (s, 2 H), 3.09 (t, J = 5.8 Hz, 2 H), 2.76 (t, J = 5.8 Hz, 2 H). 13C NMR (151 MHz, DMSO-d6) δ 161.7 (Cq), 157.6 (Cq), 157.3 (Cq), 156.7 (Cq, d, JCF = 246.8 Hz), 150.1 (CH), 145.1 (Cq), 127.6 (CH, d, JCF = 1.7 Hz), 126.7 (CH, d, JCF = 8.3 Hz), 126.6 (Cq), 124.2 (CH, d, JCF = 3.5 Hz), 121.2 (CH), 115.7 (CH, d, JCF = 20.2 Hz), 112.9 (Cq), 42.3 (CH2), 42.2 (CH2), 31.6 (CH2). QC purity (UV at 215 nm) > 99.5%.
In vitro biological activity
Cell viability assay
Human cancer cell lines A549 (lung adenocarcinoma, ATCC CCL-185), DU-145 (androgen-independent prostate cancer, ATCC HTB-81), MCF7 (breast adenocarcinoma, ATCC HTB-22) and HeLa (cervical carcinoma, ATCC CCL-2) were obtained from ATCC. Human melanoma cell lines A375 (88113005) were obtained from European Collection of Authenticated Cell Cultures (ECACC).
Cells were routinely grown in minimal essential medium containing Eagle’s salts and L-glutamine (A549, DU-145, MCF7, and HeLa) or Dulbecco’s Modified Minimal Essential Medium (A375) supplemented with 10% heat-inactivated fetal bovine serum (FBS) in a humidified atmosphere of 5% CO2 at 37 °C. To assess the antiproliferative activity of the compounds, cells were seeded at a density of 2500 cells/well (HeLa), 5000 cells/well (A549, DU145, and A375) or 10,000 cells/well (MCF7) in 96-well plates, and cell viability was measured using the MTT assay. Values are reported as the mean ± SD of two independent experiments.
Topoisomerase Inhibition
0.125 µg of pBR322 (Inspiralis) were incubated with increasing concentrations (0.5–200 µM) of tested compounds for 1 h at 37 °C in the presence/absence of 1 U of human topoisomerase IIα (Inspiralis) in the required buffer (1X). Reaction products were resolved on a 1% agarose gel prepared in 1X TAE (10 mM Tris 1 mM EDTA, 0.1% acetic acid, pH 8.0). After the electrophoretic run (5 V/cm for about 90 min) the DNA bands were visualized by ethidium bromide staining, photographed and quantified using a Geliance 2000 apparatus.
In vitro DMPK evaluation
In vitro metabolic stability
10 mM DMSO stock solution of test compound was pre-incubated at 37˚C for 15 min with mouse liver microsomes in 0.1 M Tris-HCl buffer (pH 7.4) with 10% DMSO. The final concentration was 4.6 µM (0.1% DMSO). After pre-incubation, the co-factors (NADPH, G6P, G6PDH and MgCl2 pre-dissolved in 0.1 M Tris-HCl) were added to the incubation mixture and the incubation was continued at 37˚C for 1 h. At each time point (0, 5, 15, 30, 60 min), 30 µL of incubation mixture was diluted with 200 µL cold MeCN spiked with 200 nM of an appropriate internal standard, followed by centrifugation at 3.270 x g for 15 min. The supernatant was further diluted (1:1) with H2O for analysis. A reference incubation mixture (microsomes without cofactors) was prepared for each test compound and analyzed at t = 0 and 60 min in order to verify the compounds stability in the matrix. The two time points were diluted as for the time points of the incubation mixture above. The concentration of test compound was quantified by LC-MS/MS on a Waters ACQUITY UPLC-MS/MS system consisting of a triple quadrupole detector (TQD) mass spectrometer equipped with an electrospray ionization interface (ESI) and a photodiode array detector (PDA) from Waters Inc. (Milford, MA, USA). The analyses were run on an ACQUITY UPLC BEH C18 (50 × 2.1 mmID, particle size 1.7 μm) with a VanGuard BEH C18 pre-column (5 × 2.1 mmID, particle size 1.7 μm) at 40 °C, using 0.1% HCOOH in H2O (A) and 0.1% HCOOH in MeCN (B) as mobile phase. Electrospray ionization was applied in positive mode. Compound-dependent parameters as MRM transitions and collision energy were developed for each compound. The percentage of test compound remaining at each time point relative to t = 0 was calculated by the response factor on the basis of the internal standard peak area. The percentage of test compound versus time was plotted and fitted by GraphPad Prism (GraphPad Software, Version 5 for Windows, CA, USA, www.graphpad.com) to estimate the compounds half-life (t½) which was reported as mean value along with the standard deviation (n = 3).
In vitro plasma stability
10 mM DMSO stock solution of test compound was diluted 50-fold with DMSO-H2O (1:1) and incubated at 37˚C for 2 h with mouse plasma added 5% DMSO (pre-heated at 37˚C for 10 min). The final concentration was 2 µM (0.5% DMSO). At each time point (0, 5, 15, 30, 60, 120 min), 50 µL of incubation mixture was diluted with 200 µL cold MeCN spiked with 200 nM of internal standard, followed by centrifugation at 3.500 x g for 20 min. The supernatant was further diluted (1:1) with H2O for analysis. The concentration of test compound was quantified by LC-MS/MS on a Waters ACQUITY UPLC-MS/MS system as defined above. The response factors, calculated on the basis of the internal standard peak area, were plotted over time. When possible, response vs. time profiles were fitted with Prism (GraphPad Software, Inc., USA) to estimate compounds half-life in plasma.
Aqueous kinetic solubility
The aqueous kinetic solubility was determined from a 10 mM DMSO stock solution of test compound in Phosphate Buffered Saline (PBS) at pH 7.4. The study was performed by incubation of an aliquot of 10 mM DMSO stock solution in PBS (pH 7.4) at a target concentration of 250 µM resulting in a final concentration of 2.5% DMSO. The incubation was carried out under shaking at 25 °C for 24 h followed by centrifugation at 21.100 x g for 30 min. The supernatant was further diluted (4:1) in MeCN and analyzed by LC-MS for the quantification of dissolved compound by UV at 215 nm. The analyses were performed on a Waters ACQUITY UPLC-MS system consisting of a single quadrupole detector (SQD) mass spectrometer equipped with an electrospray ionization interface (ESI) and a photodiode array detector (PDA) from Waters Inc. (Milford, MA, USA). The analyses were run on an ACQUITY UPLC BEH C18 column (50 × 2.1 mmID, particle size 1.7 μm) with a VanGuard BEH C18 pre-column (5 × 2.1 mmID, particle size 1.7 μm), using 10 mM NH4OAc in H2O at pH 5 adjusted with AcOH (A) and 10 mM NH4OAc in Me-H2O (95:5) at pH 5 (B) as mobile phase. The aqueous kinetic solubility (in µM) was calculated by dividing the peak area of dissolved test compound (supernatant) and test compound in the reference (250 µM of test compound in MeCN) and multiply by the target concentration and dilution factor.
Thermodynamic solubility
The thermodynamic solubility was determined by addition of Phosphate Buffered Saline (PBS) at pH 7.4 to an excess of solid test compound. The study was performed by incubation of an aliquot of 2.5 mg of test compound in 500 µL of PBS at pH 7.4. The suspension was shaken at 300 rpm for 24 h at 25 °C. At the end of the incubation, the saturated solution was filtered. The filtrate was further diluted with MeCN and then analyzed by LC-MS for the quantification of dissolved compound (in µM) by UV at a specific wavelength (λmax of the test compound). The analyses were performed on a Waters ACQUITY UPLC-MS system as defined above. The test compound was quantified towards a calibration curve in MeCN-H2O (1:1).
Molecular Docking
Molecular docking was performed at three distinct binding sites: the DNA cleavage site and the ATP-binding pocket at the N-terminal ___domain of TopoII, in the presence and absence of the metal cofactor Mg²⁺. For the DNA cleavage site, the structure from PDB entry 5GWK (resolution 3.15 Å)24 was used, while for exploring the ATP-competitive mechanism, PDB entry 1ZXM (resolution 1.87 Å)25 was employed. The structures were refined using the Protein Preparation Wizard workflow in Maestro (Release 2021-3)26. Specifically, missing side chains and loops were completed with Prime27, hydrogen atoms were added, and charges and protonation states were assigned by titrating the protein at physiological pH. Steric clashes were minimized through a few refinement steps until the RMSD of non-hydrogen atoms reached 0.30 Å using the OPLS3 force field28. The compounds were prepared using LigPrep software within Maestro. First, hydrogens were added, and ionization states at physiological pH were generated. Then, tautomers and all stereochemical isomers were created. A cubic grid box of 26 × 26 × 26 ų was centered on etoposide and the native ATP ligand. Finally, molecular docking was performed using Glide29,30,31 with the Extra Precision (XP) scoring function, rewarding the planarity of conjugated π-groups, sampling nitrogen inversions, adding an energy term to account for aromatic hydrogen bonds, and generating a maximum of 15 poses. All other parameters were set to their default values.
Data availability
Data is provided within the manuscript or supplementary information files. The data that support the findings of this study are available from the corresponding author, M.D.V.
References
Deweese, J. E., Osheroff, M. A. & Osheroff, N. D. N. A. Topology and topoisomerases: teaching a ‘knotty’ subject. Biochem. Mol. Biol. Educ. 37, 2–10 (2008).
Nitiss, J. L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer. 9, 327–337 (2009).
Vos, S. M., Tretter, E. M., Schmidt, B. H. & Berger, J. M. All tangled up: how cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell. Biol. 12, 827–841 (2011).
Pommier, Y., Sun, Y., Huang, S. N. & Nitiss, J. L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell. Biol. 17, 703–721 (2016).
Ashley, R. E. & Osheroff, N. Knots, Low-Dimensional Tolopogy and Apllications (Eds Colin C. Adams Et Al). (Springer International Publishing).
Deweese, J. E. & Osheroff, N. The DNA cleavage reaction of topoisomerase II: Wolf in sheep’s clothing. Nucleic Acids Res. 37, 738–748 (2009).
Pommier, Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 8, 82–95 (2013).
Nitiss, J. L. & Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer. 9, 338–350 (2009).
Joannides, M. et al. Molecular pathogenesis of secondary acute promyelocytic leukemia. Mediterr. J. Hematol. Infect. Dis. 3, e2011045 (2011).
Pendleton, M., Lindsey, R. H., Felix, C. A., Grimwade, D. & Osheroff, N. Topoisomerase II and leukemia. Ann. N Acad. Sci. 1310, 98–110 (2014).
Bailly, C. Contemporary challenges in the design of topoisomerase II inhibitors for Cancer chemotherapy. Chem. Rev. 112, 3611–3640 (2012).
Palermo, G. et al. An optimized polyamine moiety boosts the potency of human type II topoisomerase poisons as quantified by comparative analysis centered on the clinical candidate F14512. Chem. Commun. 51, 14310–14313 (2015).
Hu, W. et al. Discovery of novel topoisomerase II inhibitors by medicinal chemistry approaches. J. Med. Chem. 61, 8947–8980 (2018).
Palermo, G., Stenta, M., Cavalli, A., Dal Peraro, M. & De Vivo, M. Molecular simulations highlight the role of metals in catalysis and Inhibition of type II topoisomerase. J. Chem. Theory Comput. 9, 857–862 (2013).
Palermo, G. et al. Catalytic metal ions and enzymatic processing of DNA and RNA. Acc. Chem. Res. 48, 220–228 (2015).
Minniti, E. et al. Novel xanthone-polyamine conjugates as catalytic inhibitors of human topoisomerase IIα. Bioorg. Med. Chem. Lett. 27, 4687–4693 (2017).
Ortega, J. A. et al. Pharmacophore hybridization to discover novel topoisomerase II poisons with promising antiproliferative activity. J. Med. Chem. 61, 1375–1379 (2018).
Oviatt, A. A. et al. Polyamine-containing Etoposide derivatives as poisons of human type II topoisomerases: differential effects on topoisomerase IIα and IIβ. Bioorg. Med. Chem. Lett. 28, 2961–2968 (2018).
Riccardi, L., Genna, V. & De Vivo, M. Metal–ligand interactions in drug design. Nat. Rev. Chem. 2, 100–112 (2018).
Arencibia, J. M. et al. Biochemical characterization, and in vivo pharmacokinetics studies of novel topoisomerase II poisons with promising antiproliferative activity. J. Med. Chem. 63, 3508–3521 (2020). Design, Synthesis, Dynamic Docking.
Ortega, J. A. et al. Novel, potent, and druglike tetrahydroquinazoline inhibitor that is highly selective for human topoisomerase II α over β. J. Med. Chem. 63, 12873–12886 (2020).
Sarogni, P. et al. Tumor growth-arrest effect of tetrahydroquinazoline-derivative human topoisomerase II-alpha inhibitor in HPV-negative head and neck squamous cell carcinoma. Sci. Rep. 14, 9150 (2024).
De Vivo, M., Masetti, M., Bottegoni, G. & Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 59, 4035–4061 (2016).
Wang, Y. R. et al. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt(II)-methionine coordination chemistry. Nucleic Acids Res. 45, 10861–10871 (2017).
Wei, H., Ruthenburg, A. J., Bechis, S. K. & Verdine, G. L. Nucleotide-dependent ___domain movement in the ATPase ___domain of a human type IIA DNA topoisomerase. J. Biol. Chem. 280, 37041–37047 (2005).
Madhavi Sastry, G., Adzhigirey, M., Day, T., Annabhimoju, R. & Sherman, W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234 (2013).
Jacobson, M. P. et al. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinforma. 55, 351–367 (2004).
Harder, E. et al. OPLS3: A force field providing broad coverage of Drug-like small molecules and proteins. J. Chem. Theory Comput. 12, 281–296 (2016).
Friesner, R. A. et al. Glide: A new approach for rapid, accurate Docking and scoring. 1. Method and assessment of Docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Halgren, T. A. et al. Glide: A new approach for rapid, accurate Docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759 (2004).
Friesner, R. A. et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for Protein – Ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Acknowledgements
C.S thanks the European Union-Next GenerationEU (PNRR M4C2-Investimento 1.4- CN00000041). M.D.V. also acknowledges the project “National Center for Gene Therapy and Drug based on RNA Technology” (CN00000041). Financed by NextGenerationEU PNRR MUR – M4C2 – Action 1.4- Call. “Potenziamento strutture di ricerca e di campioni nazionali di R&S” (CUP: J33C22001130001). Also, M. D. V. thanks AIRC for financial support (IG 30631). We thank S. Venzano for the preparation of the plates with the compounds’ solution for the screening.
Author information
Authors and Affiliations
Contributions
J.A.O. and M.B. designed and synthesized the compounds; J.M.A. and M.N. performed the viability assay; M.A.L.S. performed molecular modeling studies; S.M.B. and A.A. carried out DMPK properties analysis and QC; C.S. and M.D.V conceived the study, supervised the work and wrote the manuscript with the help of N.B and J.A.O. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare the following competing financial interest(s): M.D.V., J.A.O, J.M.A, and C.S. are coinventors on patent related to this work (WO2018/114700Al). All other authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Brindani, N., La Serra, M.A., Ortega, J.A. et al. Tetrahydropyrido[4,3-d]pyrimidines as a new active scaffold for human topoisomerase II inhibitors. Sci Rep 15, 19618 (2025). https://doi.org/10.1038/s41598-025-04773-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-04773-z
