
Abstract
This study successfully synthesized a magnetically recoverable ZnFe₂O₄@g-C₃N₄ heterojunction photocatalyst by anchoring ZnFe₂O₄ nanoparticles (20–30 nm) onto a mesoporous g-C₃N₄ framework via a hydrothermal method. Comprehensive characterizations, including XRD, SEM, TEM, and UV–Vis spectroscopy, confirmed the formation of a porous multilayer structure with uniform dispersion of ZnFe₂O₄ nanoparticles on the g-C₃N₄ surface. Tight interfacial heterojunction bonding significantly enhanced photogenerated charge separation. BET analysis revealed a high specific surface area ( 855.9 m2/g) due to the mesoporous architecture, while TEM further elucidated efficient electron transport at the heterojunction interface. Under visible light irradiation, the composite achieved complete degradation of methylene blue (MB) through synergistic effects of extended light absorption, accelerated interfacial charge transfer, and high-density active sites. At an optimal ZnFe₂O₄ loading of 59.1 wt%, the degradation efficiency reached 99.99% within 40 min, with a rate constant (0.253 min⁻1) ninefold higher than that of pristine g-C₃N₄. The introduction of H₂O₂ activated a photo-Fenton mechanism, further boosting hydroxyl radical (·OH) generation and improving degradation efficiency by 12 times. Additionally, the inherent ferromagnetism of ZnFe₂O₄ enabled facile magnetic recovery, with catalytic activity retention exceeding 95% after 10 consecutive cycles. The ZnFe₂O₄@g-C₃N₄ heterojunction photocatalyst developed in this work integrates high degradation efficiency, magnetic recyclability, and structural stability, demonstrating significant potential for industrial wastewater treatment and environmental remediation. This study provided a novel strategy for designing sustainable photocatalytic systems, offering insights into dual-mechanism (photocatalytic/Fenton-like) synergies and scalable heterojunction engineering for advanced pollutant degradation.
Similar content being viewed by others
Introduction
Water is an indispensable resource for sustaining life across ecosystems, yet its contamination by antibiotics, pesticides, and synthetic dyes has escalated into a global environmental crisis, jeopardizing human health and ecological stability. Addressing this challenge requires innovative solutions to degrade persistent pollutants effectively. Advanced oxidation processes (AOPs), particularly those leveraging visible-light-driven photocatalysis and Fenton-like reactions, have emerged as a frontier technology for water purification due to their capacity to generate highly reactive oxygen species (ROS) under mild conditions1,2,3,4. Central to these processes is the development of efficient, stable, and recyclable heterojunction catalysts capable of optimizing charge separation and ROS generation.
Recent advancements highlight the potential of multiphase composite catalysts, where materials with distinct band structures synergize to enhance charge carrier dynamics. For instance, high-entropy catalysts with tailored conduction and valence band positions facilitate rapid separation of photogenerated electron–hole pairs, amplifying catalytic efficiency5,6,7,8,9,10. Mesoporous materials, in particular, serve as ideal substrates due to their high surface area and adsorption capacity. When functionalized with metals or metal oxides, they promote electron transfer and activate H₂O₂ decomposition into hydroxyl radicals (·OH), a critical step in pollutant degradation5,6,7,8,9,10.
Zinc ferrite (ZnFe₂O₄, ZFO), a narrow-bandgap (1.0–2.0 eV) n-type semiconductor, has garnered attention for its visible-light absorption, magnetic recoverability, and photo-fenton effect, which synergize with H₂O₂ to generate ROS11,12,13,14,15,16,17,18,19,20,21. Concurrently, graphitic carbon nitride (g-C₃N₄), a metal-free polymeric semiconductor with a 2.7 eV bandgap and redox-active bands (−1.3 and + 1.4 eV), offers tunable structural properties, high chemical stability, and enhanced charge migration via its 2D/3D architectures22,23,24,25. Integrating ZFO with g-C₃N₄ presents a promising strategy to bridge their complementary advantages26,27,28,29,30,35,36,37,38,39,40,41: First, ZnFe₂O₄, as a narrow-bandgap spinel ferrite semiconductor, possesses a complementary band structure to g-C₃N₄. This enables the construction of Type-II or Z-scheme heterojunctions, where the built-in electric field at the interface facilitates directional migration and efficient separation of photogenerated electron–hole pairs (e⁻-h⁺), thereby drastically reducing recombination rates26,27,28,29,30. Second, the uniform loading of ZnFe₂O₄ nanoparticles onto g-C₃N₄ nanosheets or within mesopores forms core–shell or layered architectures. This not only enhances active site density and shortens charge transfer pathways but also optimizes mass transport kinetics35,36,37,38,39,40,41. Third, the composite system broadens the light absorption spectrum: g-C₃N₄ dominates the visible region, while ZnFe₂O₄ extends absorption to near-infrared and partial visible light, achieving full-spectrum solar energy utilization up to 700 nm9,10,11. Furthermore, the high conductivity of ZnFe₂O₄ provides rapid electron transport channels for g-C₃N₄, suppressing bulk charge recombination. As a protective layer, ZnFe₂O₄ mitigates photocorrosion of g-C₃N₄ and strengthens interfacial bonding via covalent bonds or hydrogen bonds, endowing the material with superior chemical stability compared to the weak van der Waals interactions in pure g-C₃N₄. ZFO’s magnetic recyclability and ROS-generating capacity, coupled with g-C₃N₄’s expansive surface area and charge transport efficiency. In summary, the synergistic optimization of light absorption, carrier separation efficiency, structural stability, and anti-photocorrosion properties positions this composite as an innovative strategy for high-efficiency solar-driven catalytic systems.
In this study, mesoporous g-C₃N₄ was synthesized via thermal polymerization of urea and melamine, followed by the hydrothermal deposition of ZFO nanoparticles using zinc acetate and iron chloride precursors. The resultant ZnFe₂O₄@g-C₃N₄ heterostructure was systematically characterized through SEM, TEM, XRD, PL and UV–Vis spectroscopy to elucidate its structural, morphological, and optoelectronic properties. The photocatalytic performance was evaluated via visible-light-driven degradation of methylene blue (MB), with mechanistic insights into charge transfer pathways and ROS generation explored. Furthermore, the magnetic recoverability and cyclic stability of the composite were assessed to underscore its practical viability. This work not only advanced the design of multifunctional heterojunction catalysts but also highlighted their potential in environmental remediation, particularly for industrial wastewater treatment and water quality restoration.
Experimental section
Reagents and characterization
All chemicals, including urea (CH₄N₂O, ≥ 99%), melamine (C₃H₆N₆, ≥ 99%), zinc acetate dihydrate (Zn(CH₃COO)₂·2H₂O, ≥ 99%), iron(III) chloride hexahydrate (FeCl₃·6H₂O, ≥ 98%), methylene blue (C₁₆H₁₈ClN₃S, ≥ 95%), and hydrogen peroxide (H₂O₂, 30 wt%), were of analytical grade and procured from Sinopharm Chemical Reagent Co., Ltd. (China). Deionized water (resistivity ≥ 18.2 MΩ·cm) was produced using a laboratory-grade purification system.
The crystallographic properties of the synthesized g-C₃N₄ and ZnFe₂O₄@g-C₃N₄ composites were analyzed using an Ultima IV X-ray diffractometer (Rigaku Corporation, Japan) with Cu-Kα radiation (λ = 1.5406 Å) operated at 40 kV and 40 mA. Morphological and microstructural features were examined using a JEM-2100 F field-emission transmission electron microscope (JEOL Ltd., Japan) at an accelerating voltage of 200 kV. The optical absorption properties and photocatalytic degradation kinetics of methylene blue were monitored via a PerkinElmer LAMBDA750 UV/Vis/NIR spectrophotometer, with spectra recorded in the range of 200–800 nm.
Experimental process
Preparation of g-C 3 N 4 nanoparticles
Graphene-like g-C3N4 was prepared using a thermal polymerization method. Details of the sample preparation could be found in references10,11.
ZnFe 2 O 4 @g-C 3 N 4 preparation of nanoparticles
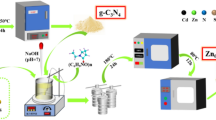
The ZnFe₂O₄@g-C₃N₄ heterostructure was synthesized via a hydrothermal route. Briefly, 0.5 g of g-C₃N₄ was uniformly dispersed in 50 mL of deionized water via ultrasonication (40 kHz, 30 min). Separately, stoichiometric ratios of zinc acetate dihydrate (Zn(CH₃COO)₂·2H₂O) and iron(III) chloride hexahydrate (FeCl₃·6H₂O) (mass ratios detailed in Table 1) were dissolved in 25 mL of deionized water under magnetic stirring (500 rpm, 30 min). The metal precursor solution was then introduced dropwise into the g-C₃N₄ suspension, followed by the addition of 0.5 g urea as a pH-modulating agent. The mixture was stirred vigorously (60 min) to ensure homogeneity.

The schematic diagram for the synthesis process of ZnFe₂O₄@g-C₃N₄.
The resultant suspension was transferred into a 100 mL polytetrafluoroethylene (PTFE)-lined autoclave and subjected to hydrothermal treatment in a programmable oven. The temperature was ramped to 150 °C at a rate of 2 °C/min and maintained for 4 h. After natural cooling to ambient temperature, the precipitate was isolated by centrifugation (7000 rpm, 15 min), followed by sequential washing with deionized water and ethanol (three cycles each) to remove residual impurities. The schematic diagram for the synthesis process of ZnFe₂O₄@g-C₃N₄ was shown in Scheme 1. The purified product was vacuum-dried at 60 °C for 24 h and stored in an airtight container for subsequent characterization and application. The samples name are list in Table 1. The symbol “−0”, “−1”, “−2”, “−3”, “−4”, “−5”, “−6” and “−7” means the increasing mole of raw material “Zn(AC)2” as 0, 0.001, 0.002, 0.003, 0.004, 0.005, 0.006, and 0.007 mol. As shown in last column, the mass ratio ZnFe2O4 to g-C3N4 and ZnFe2O4 were 0, 0.325, 0.491, 0.591, 0.658, 0.707, 0.743, and 0.771 in different samples.
Photocatalytic degradation of methylene blue
The visible-light-driven photocatalytic activity of ZnFe₂O₄@g-C₃N₄ was assessed through the degradation of methylene blue (MB) in aqueous media. Prior to illumination, 50 mg of the catalyst was dispersed in 100 mL of MB solution (30 mg/L) within a 100 mL borosilicate glass reactor under darkroom conditions. The suspension was ultrasonicated (40 kHz, 3 min) to ensure uniform catalyst distribution, followed by magnetic stirring (60 min) to establish adsorption–desorption equilibrium between the dye and catalyst surface. The initial pH was one of important parameters in photocatalytic processes. During all the degradation experiments, the pH of the methylene blue aqueous solution, which was the target for degradation, was adjusted to pH = 7 using dilute solutions of sodium hydroxide and hydrochloric acid.
A 300 W xenon lamp equipped with a 420 nm cutoff filter was positioned 20 cm above the reactor to simulate visible-light irradiation. During all the degradation experiments, the temperature of the methylene blue aqueous solution, which was the target for degradation, was adjusted and maintained to 25℃ using condensate system. Aliquots (3 mL) were extracted at 10-min intervals and immediately centrifuged (7000 rpm, 2 min) to isolate the catalyst, thereby preventing interference in absorbance measurements. The residual MB concentration was quantified via UV–Vis spectroscopy by monitoring the attenuation of its characteristic absorption band at λ = 664 nm. The schematic diagram for degrading MB was shown in Scheme 2. The degradation efficiency (η, %) and real time change of absorption intensity were calculated using the equation:
where C0 and C represent the initial and time-dependent absorbance intensities of MB, respectively10,11.

The schematic diagram for degrading MB.
Results and discussion
XRD structural characterization
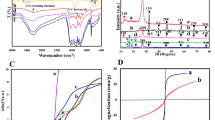
The crystallographic structures of pristine g-C₃N₄ and the ZnFe₂O₄@g-C₃N₄ composite were analyzed via X-ray diffraction (XRD). As illustrated in Fig. 1, the XRD pattern of ZnFe₂O₄@g-C₃N₄ exhibits distinct diffraction peaks corresponding to both g-C₃N₄ and ZnFe₂O₄ phases, confirming the coexistence of the two components. For pristine g-C₃N₄, two characteristic peaks were observed at 13.1° and 27.5°, indexed to the (001) and (002) crystallographic planes, respectively. These reflections arose from the in-plane structural packing of tri-s-triazine units and the interlayer stacking of conjugated aromatic systems, consistent with the typical graphitic carbon nitride framework (JCPDS 87–1526)10,11.

The XRD patterns of samples g-C3N4 and ZnFe2O4@g-C3N4.
In the composite, additional diffraction peaks at 2θ values of 18°, 30.2°, 35.6°, 36.9°, 43.3°, 45°, 53.7°, 57.3°, and 62.8° were assigned to the (111), (220), (311), (222), (400), (331), (422), (511), and (440) planes of cubic spinel ZnFe₂O₄ (JCPDS 01–070–6490)12,13. Notably, these peaks displayed slight angular deviations compared to the standard reference values. These systematic peak shifts were attributed to lattice distortion in ZnFe₂O₄ induced by interfacial electronic interactions at the heterojunction with g-C₃N₄. Such lattice parameter modifications—calculated via Bragg’s law (nλ = 2 dsinθ) to reflect a unit cell contraction—provided definitive evidence of strong electronic coupling between ZnFe₂O₄ and g-C₃N₄, ruling out mere physical mixing. The coexistence of these distinct diffraction features confirmed the successful formation of a heterostructure, where ZnFe₂O₄ nanoparticles were anchored onto the g-C₃N₄ matrix without disrupting its layered architecture. These results validated the effective synthesis of a phase-pure ZnFe₂O₄@g-C₃N₄ composite, laying a foundation for its enhanced photocatalytic functionality.
The heterojunction ZnFe₂O₄@g-C₃N₄ photocatalysts were well synthesized and crystallized in nanomaterials. The size of ZnFe₂O₄ nanoparticles which loaded on the surface of the g-C3N4nanosheets were calculated according the Scherrer equation26:
where the parameter D, k, λ, β and θ were defined as particle size, Scherrer constant (0.89), X-ray wavelength (0.15418 nm), full width at half maximum and angle at maximum, respectively. The size of the ZnFe₂O₄ on the g-C3N4 were calculated as 22.31 nm and 21.13 nm according the characteristic peak which located at the crystal planes (113) of red and blue curves in Fig. 1. The calculated particle size of ZnFe₂O₄ (22.31 nm) nanoparticles distributed on the g-C₃N₄ surface aligns with the SEM (20–30 nm) and TEM (10–27.5 nm) analysis results, confirming structural consistency between theoretical models and experimental observations.
The blue curve represented the used ZnFe₂O₄@g-C₃N₄ in the photocatalytic degradation process. Throughout the experiment, the diffraction peaks remained essentially unchanged, with the exception of their intensity. The consistent diffraction peak positions before and after the reaction signify that the crystal structures of g-C₃N₄ and ZnFe₂O₄ were unmodified. The attenuation of ZnFe₂O₄'s diffraction peaks could likely be attributed to variations in preferred orientation, structural impairment caused by photocorrosion, or modifications in content and dispersibility. Conversely, g-C₃N₄ exhibited relatively high stability, leading to negligible changes in its peak intensity.
Morphological and microstructural analysis
The morphological evolution and interfacial characteristics of pristine g-C₃N₄, ZnFe₂O₄ and the ZnFe₂O₄@g-C₃N₄ composite were investigated via scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As depicted in Fig. 2(a), the thermal polymerization of urea and melamine yielded g-C₃N₄ with a lamellar morphology characterized by pronounced wrinkling and sheet-like stacking, a hallmark of its graphitic architecture. The ZnFe₂O₄ were approximate cube particle and diameter dispersed from 85 to 120 nm in Fig. 2(b) and 2(d). Upon integration with ZnFe₂O₄ (Fig. 2c), the composite retained the layered framework of g-C₃N₄ but exhibited reduced lateral dimensions of the individual sheets, likely due to fragmentation during hydrothermal processing. Crucially, ZnFe₂O₄ nanoparticles (20–30 nm) were uniformly dispersed across the g-C₃N₄ matrix without agglomeration, confirming the successful formation of an intimate heterointerface between the two phases. This structural synergy was anticipated to enhance charge carrier separation and surface reactivity, thereby augmenting photocatalytic efficiency.

The SEM images of samples g-C3N4, ZnFe2O4 and ZnFe2O4@g-C3N4.
Further insights into the microstructural features were obtained through TEM (Fig. 3(a)–(d)). Pristine g-C₃N₄ displayed a highly porous, crumpled 2D structure with abundant in-plane voids (Fig. 3(a)), attributed to the release of gaseous byproducts NH₃ during thermal condensation. These mesopores (5–15 nm diameter) not only increase the material’s specific surface area but also facilitate reactant diffusion and active site accessibility. The ZnFe₂O₄ were polycrystalline structure and diameter dispersed from 80 to 100 nm in Fig. 3(b). In the ZnFe₂O₄@g-C₃N₄ composite (Fig. 3(c)), the g-C₃N₄ sheets maintained their layered topology, while ZnFe₂O₄ nanoparticles were anchored uniformly across the surface. As shown in Fig. 3(d), the diameter of ZnFe₂O₄ dispersed 10 to 27.5 nm. Upon analyzing the reason for the reduction in the particle size of ZnFe₂O₄, it was mainly attributed to the fact that the porous and curled structures of g-C₃N₄ restricted the growth of ZnFe₂O₄. Consequently, compared to pure ZnFe₂O₄ particles, the particle size was notably smaller.

The TEM images of samples g-C3N4, ZnFe2O4 and ZnFe2O4@g-C3N4.
High-resolution TEM (Fig. 4) revealed lattice fringes corresponding to the (220) and (311) plane of cubic ZnFe₂O₄ and interplanar spacing was 0.2787 and 0.2426 nm, corroborating the coexistence of both phases without interfacial contamination. The absence of particle aggregation and the coherent integration of ZnFe₂O₄ within the g-C₃N₄ scaffold underscored the efficacy of the synthesis strategy in fostering a well-defined heterostructure, a critical determinant of enhanced catalytic performance.

The HRTEM images of ZnFe2O4@g-C3N4.
Nitrogen adsorption–desorption analysis
The textural properties of the ZnFe₂O₄@g-C₃N₄ composite were evaluated through nitrogen adsorption–desorption measurements. As depicted in Fig. 5(a), the composite exhibited a type IV isotherm with an H3 hysteresis loop, characteristic of mesoporous materials featuring slit-like pores formed by platelet aggregates. A sharp uptake at high relative pressure (P/P₀ > 0.95) signified the coexistence of macropores, attributed to the volatilization of ammonia gas during the thermal polymerization of g-C₃N₄ precursors. The Brunauer–Emmett–Teller (BET) specific surface area of the g-C₃N₄ was calculated as 855.9 m2·g⁻1, with reduced surface areas of 654.2, 358.4, 37.9, 29.2, 23.2, 20.22, and 14.1 m2·g⁻1 observed under increased ZnFe₂O₄. According to nitrogen adsorption–desorption analysis, the ZnFe₂O₄@g-C₃N₄ composite exhibited a significant reduction in specific surface area with increasing ZnFe₂O₄ loading. This decrease can be attributed to the partial occupation of the porous g-C₃N₄ framework by ZnFe₂O₄ nanoparticles. However, the formation of a well-defined ZnFe₂O₄/g-C₃N₄ heterojunction became more prominent at higher ZnFe₂O₄ content, as evidenced by the concomitant rise in catalytically active sites. While the pore-blocking effect of ZnFe₂O₄ diminished the surface area, the enhanced interfacial charge transfer and increased density of reactive sites at the heterojunction interface counterbalanced this limitation. The interplay between these competing factors—surface area reduction and active site enrichment—culminated in the ZnFe₂O₄@g-C₃N₄−3 composite demonstrating optimal catalytic performance, highlighting the critical role of heterojunction engineering in optimizing activity despite structural trade-offs.

The absorption curves of samples ZnFe2O4@g-C3N4.
Pore size distribution derived from the Barrett-Joyner-Halenda (BJH) model (Fig. 5(b)) reveals a bimodal architecture dominated by mesopores (2.5–10 nm, average diameter: 3.4–5.6 nm) and subsidiary macropores (10–100 nm). The mesopores originate from interparticle voids within the aggregated g-C₃N₄ layers, while macropores arose from gas evolution during synthesis. Such hierarchical porosity facilitated efficient mass transport of reactants and access to catalytic sites, synergistically enhancing degradation kinetics. These structural attributes—high surface area, dual-scale porosity, and interfacial connectivity—underscored the ZnFe₂O₄@g-C₃N₄ composite’s suitability for advanced photocatalytic applications.
Surface elemental and chemical state analysis
The chemical composition and interfacial interactions within the ZnFe₂O₄@g-C₃N₄ heterostructure were probed via X-ray photoelectron spectroscopy (XPS). The survey spectrum (Fig. 6(a)) confirms the coexistence of oxygen (O), nitrogen (N), carbon (C), zinc (Zn), and iron (Fe), corroborating the composite’s hybrid nature. High-resolution spectra were deconvoluted to elucidate bonding configurations and oxidation states. In O 1 s Spectrum (Fig. 6(b)), the O 1 s envelope resolves into four components at 529.75, 530.95, 531.45, and 531.95 eV. Peaks at 529.75 and 530.95 eV are attributed to Zn–O–Fe coordination in the spinel ZnFe₂O₄ lattice, while contributions at 531.45 and 531.95 eV correspond to surface-adsorbed hydroxyl (–OH) and carboxyl (–HO–C = O) groups, indicative of oxygen-containing functional moieties. In N 1 s Spectrum (Fig. 6(c)), the N 1 s profile exhibits three distinct binding energies at 398.55, 399.00, and 399.55 eV. The dominant peak at 398.44 eV arises from C–N–C coordination in the tri-s-triazine units of g-C₃N₄, while contributions at 399.00 and 399.55 eV correspond to sp2-hybridized nitrogen (N–(C)₃) and terminal amino groups (–NH₂), respectively, confirming the integrity of the g-C₃N₄ framework. In C 1 s Spectrum (Fig. 6(d)), deconvolution of the C 1 s signal reveals four components: (i) adventitious carbon (C = C/C–C, 284.95 eV), (ii) Zn–C bonds (284.55 eV) associated with oxygen vacancies, (iii) graphitic sp2 carbon (285.45 eV), and (iv) a prominent peak at 288.1 eV assigned to N–C = N coordination in the aromatic triazine rings of g-C₃N₄. In Fe 2p Spectrum (Fig. 6(e)), the Fe 2p region displays spin–orbit doublets for Fe 2p₃/₂ (710.56 eV) and Fe 2p₁/₂ (724.66 eV), accompanied by a satellite peak at 718.96 eV. The Fe 2p₃/₂ peak splits into two components at 710.56 eV (Fe3⁺ in octahedral sites) and 712.36 eV (Fe3⁺ with ligand field effects), confirming the dominance of Fe3⁺ in the ZnFe₂O₄ lattice. The absence of Fe2⁺ signatures (typically < 709 eV) underscores the stability of the spinel structure. In Zn 2p Spectrum (Fig. 6(f)), the Zn 2p spectrum exhibits symmetric peaks at 1021.92 eV (Zn 2p₃/₂) and 1045.02 eV (Zn 2p₁/₂), characteristic of Zn2⁺ in tetrahedral coordination within ZnFe₂O₄. The 23.1 eV spin–orbit splitting further validates the + 2 oxidation state of Zn. The XPS analysis confirms the coexistence of ZnFe₂O₄ and g-C₃N₄ phases with interfacial Zn–C and Fe–O–N bonding, indicative of strong electronic coupling. This synergy, coupled with oxygen vacancies and surface functional groups, underpins the composite’s enhanced photocatalytic activity.

X-ray photoelectron spectrum of ZnFe2O4@g-C3N4.
Optical property
The optical absorption characteristics of g-C₃N₄ and ZnFe₂O₄@g-C₃N₄ nanocomposites were analyzed using ultraviolet–visible diffuse reflectance spectroscopy (UV–vis DRS). As illustrated in Fig. 7(a), pure g-C₃N₄ exhibited an absorption edge within the visible light spectrum. Upon incorporation of ZnFe₂O₄ nanoparticles onto the g-C₃N₄ surface, the composite demonstrated a pronounced red shift in its absorption edge, accompanied by a significant enhancement in absorption intensity across the visible to near-infrared (Vis–NIR) regions. This shift indicates extended light-harvesting capabilities. The optical band gaps (Eg) of the synthesized photocatalysts were determined via the Tauc plot method, employing the equation for direct band gap semiconductors42,43,44,45:.

UV–vis–NIR diffuse reflectance spectra of ZnFe2O4@g-C3N4,(b) corresponding Tauc plots of (αhν)2 versus the photon energy (hν) of ZnFe2O4@g-C3N4.
where α, hν, Eg, and B represent the absorption coefficient, photon energy, band gap energy, and a proportionality constant, respectively. As shown in Fig. 7, the calculated Eg values for g-C₃N₄ and ZnFe₂O₄@g-C₃N₄ composites were 2.6067 eV (pure g-C₃N₄) and 2.0418 eV, 2.5994 eV, 2.5611 eV, 2.5282 eV, 1.8976 eV, 1.8926 eV, and 1.8903 eV (ZnFe₂O₄@g-C₃N₄), respectively. The progressive reduction in Eg with increasing ZnFe₂O₄ content confirmed band structure modulation. The UV–vis-NIR DRS spectra further revealed that the formation of a p-n heterojunction between g-C₃N₄ and ZnFe₂O₄ markedly improved optical absorption under Vis–NIR irradiation. This enhanced absorption range promotes more efficient utilization of solar energy compared to pristine g-C₃N₄, suggested superior photocatalytic activity for the ZnFe₂O₄@g-C₃N₄ nanocomposites in Vis–NIR regions.
Photoluminescence (PL) spectroscopy was employed to evaluate the electron–hole separation efficiency of ZnFe₂O₄@g-C₃N₄ composites in a pure aqueous system11,12,13,14,15,16. As depicted in Fig. 8, the PL spectra of the composites revealed a progressive suppression of emission peak intensity with increasing ZnFe₂O₄ content. This trend was attributed to the reduced recombination probability of photoinduced charge carriers, as PL emission originates from the radiative recombination of excited electrons and holes. The peak intensities followed the order: ZnFe₂O₄@g-C₃N₄−0 > ZnFe₂O₄@g-C₃N₄−1 > ZnFe₂O₄@g-C₃N₄−2 > ZnFe₂O₄@g-C₃N₄−3 > ZnFe₂O₄@g-C₃N₄−4 > ZnFe₂O₄@g-C₃N₄−5 > ZnFe₂O₄@g-C₃N₄−6 > ZnFe₂O₄@g-C₃N₄−7. This systematic decrease in intensity underscores the role of the ZnFe₂O₄/g-C₃N₄ heterostructure in facilitating charge separation, likely through interfacial electric field effects or staggered band alignment. Consequently, the inhibited recombination kinetics correlated with enhanced availability of free charge carriers for surface redox reactions. These results confirmed that the ZnFe₂O₄@g-C₃N₄ composites exhibited superior photocatalytic activity compared to pristine g-C₃N₄, driven by efficient electron–hole separation and prolonged carrier lifetimes under illumination.

PL spectra of samples ZnFe2O4@g-C3N4.
Photocatalytic performance
The photocatalytic activity of the ZnFe2O4@g-C3N4 heterostructure was rigorously assessed via visible-light-driven degradation of methylene blue (MB), a model organic pollutant27,28. As depicted in Fig. 9, time-resolved ultraviolet–visible (UV–Vis) spectroscopy demonstrated a monotonic decline in the characteristic absorbance intensity of MB at λ = 664 nm as a function of irradiation duration, with the signal diminishing to near-background levels after prolonged exposure. The absence of hypsochromic or bathochromic shifted in the primary absorption band, coupled with no detectable emergence of secondary absorption features within the spectral range (400–800 nm), strongly indicates the complete mineralization of MB rather than transient intermediate formation or structural isomerization. These observations unequivocally validate the robust photocatalytic efficacy of the ZnFe2O4@g-C3N4 composite, highlighting its capacity for efficient visible-light harvesting and redox-driven degradation of organic contaminants.

The changed absorption peak of MB with visible light irradiated.
The photocatalytic degradation of methylene blue (MB) by ZnFe₂O₄@g-C₃N₄ under visible light irradiation is governed by the generation of reactive oxygen species (ROS), notably hydroxyl radicals (·OH) and superoxide anions (·O₂⁻), facilitated by the heterojunction’s electronic structure27,28. As illustrated in Fig. 10(a), the degradation efficiency correlates with ZnFe₂O₄ doping concentration, achieving optimal performance at 74.3 wt% loading. Under dark conditions (Fig. 10(a), black curve), the composite exhibits substantial adsorption of MB molecules due to its mesoporous g-C₃N₄ framework, yet no degradation occurs in the absence of light. Upon visible-light irradiation, photogenerated electrons (e⁻) and holes (h⁺) are formed, with the heterojunction interface promoting efficient charge separation. This suppresses recombination, enabling prolonged ROS generation and rapid MB decomposition. Exceeding the optimal ZnFe₂O₄ loading (> 74.3 wt%) diminishes performance due to nanoparticle aggregation, which obstructs the porous g-C₃N₄ layers, reduces active site accessibility, and impedes dye-catalyst contact. Kinetic analysis (Fig. 10(c), Table 2) confirms pseudo-first-order behavior, with rate constants peaking at 74.3 wt% (0.12697 min⁻1) and declining thereafter. The synergy between g-C₃N₄’s adsorption capacity and ZnFe₂O₄’s visible-light activation is critical for maximizing degradation efficiency.

The changed catalytic performance with visible light irradiated.
The introduction of H₂O₂ (0.5 mL, 30%) amplifies catalytic activity via a photo-Fenton mechanism (Fig. 10(b) and (d)), where H₂O₂ reacts with photogenerated electrons to yield additional ·OH radicals. This dual catalytic pathway achieves complete MB degradation within 40 min, with kinetic coefficients increasing by 12-fold at 59.1 wt% ZnFe₂O₄ (Table 2). However, excessive doping (> 59.1 wt%) under H₂O₂ conditions induces shielding effects, wherein dense ZnFe₂O₄ layers hinder H₂O₂ diffusion and ROS generation, underscoring the necessity of balanced compositional design. The performance comparison between the ZnFe2O4@g-C3N4 material and reported catalysts was presented in Table 3. As is evident from the table, the photocatalytic performance of ZnFe2O4@g-C3N4 outperforms that of most of the reported photocatalysts.
A above analysis, the photocatalytic performance of the ZnFe₂O₄@g-C₃N₄ composites exhibited an optimal threshold when an equilibrium between specific surface area and active site density was achieved. During the initial reaction stage, the degradation efficiency remained limited due to the high initial dye concentration and insufficient generation of photoinduced reactive species. As the reaction progressed, the accumulation of reactive species significantly increased their collision frequency with dye molecules, thereby enhancing the degradation kinetics. Notably, the density of these reactive species was directly modulated by the ZnFe₂O₄ loading within the composite. As demonstrated in Fig. 10, the ZnFe₂O₄@g-C₃N₄−3 composite displayed superior photocatalytic activity, which originated from a synergistic effect arising from two competing factors: (i) the formation of an efficient ZnFe₂O₄/g-C₃N₄ heterojunction, which facilitated interfacial charge separation and amplified reactive species generation, and (ii) the retention of sufficient surface accessibility despite partial pore occlusion by ZnFe₂O₄ nanoparticles. This balance between heterojunction-driven electronic enhancement and structural porosity underscores the critical role of compositional optimization in maximizing photocatalytic performance.
Photocatalytic active agent
To elucidate the reactive species governing the photocatalytic degradation mechanism, radical scavenging experiments were conducted under visible light irradiation11,27,28,29,30. The active species photogenerated holes (h⁺), hydroxyl radicals (·OH), and superoxide anions (·O₂⁻) were systematically investigated using targeted quenchers: p-benzoquinone (BQ, ·O₂⁻ scavenger), tert-butanol (TBA, ·OH scavenger), and disodium ethylenediaminetetraacetate dihydrate (EDTA-2 Na, h⁺ scavenger). As depicted in Fig. 11(a), the degradation efficiency of methylene blue (MB) by ZnFe₂O₄@g-C₃N₄−3 was significantly attenuated in the presence of these inhibitors.

Elemental trapping experiment, recycled degradation rate and Vsm of catalysis.
The degradation rate declined from 99.99% (pristine system) to 81.12%, 73.11%, and 82.52% upon introduction of BQ, TBA, and EDTA-2 Na, respectively, over 40 min. The pronounced suppression of activity with TBA (73.11% remaining MB) underscores ·OH as the predominant reactive species, attributable to its potent oxidative capacity. Concurrently, the inhibition by BQ (81.12% residual MB) and EDTA-2 Na (82.52% residual MB) confirms the ancillary roles of ·O₂⁻ and h⁺ in the degradation pathway. These findings align with the heterojunction’s electronic structure, which promotes charge separation, enabling h⁺ to directly oxidize MB or react with H₂O to generate ·OH, while ·O₂⁻ forms via oxygen reduction by photogenerated electrons. This study conclusively identifies ·OH as the primary active species, with synergistic contributions from h⁺ and ·O₂⁻, underpinning the composite’s robust photocatalytic performance.
Recyclability and stability
The reusability and structural stability of the ZnFe₂O₄@g-C₃N₄−5 catalyst were evaluated through successive degradation cycles under consistent experimental conditions23,32,35. As illustrated in Fig. 11(b), the composite retained > 95% of its initial degradation efficiency after ten reuse cycles, demonstrating robust operational durability. As shown in Fig. 11(c), the material’s inherent ferromagnetism, derived from ZnFe₂O₄, enabled facile magnetic recovery using an external magnetic field, circumventing secondary pollution risks associated with conventional filtration methods. The marginal efficiency decline is attributed to partial active site deactivation via adsorbed intermediates, rather than structural degradation. These results underscore the catalyst’s viability for sustainable wastewater treatment, balancing high recyclability with minimal performance attenuation. The catalysts was recycled by Neodymium magnet after degradation of MB in Fig. 11(c). The recovery rate of the neodymium magnet-derived catalyst was only 70%, indicating a relatively low efficiency. Analytical results revealed two primary causes: first, partial destruction of the zinc ferrite (ZnFe₂O₄) crystal structure after the degradation reaction led to the loss of magnetic properties; second, the small particle size of the catalyst, combined with the viscous force of the solution post-reaction, hindered the effective separation of catalyst components. As a result, centrifugation remained the adopted method for catalyst recovery in the experiments.
Catalytic mechanism
The experimental data collectively demonstrate that the ZnFe₂O₄ − C₃N₄ heterojunction photocatalyst efficiently generates hydroxyl radicals (·OH) through the photochemical decomposition of H₂O₂ under visible light irradiation4,5,6,7,8,35,37. This process markedly enhances the separation of photogenerated electron–hole pairs while suppressing charge recombination, as evidenced by the progressive decline in photoluminescence (PL) intensity with increasing ZnFe₂O₄ loading on the g-C₃N₄ surface (Fig. 8). The formation of heterojunction between ZnFe₂O₄ nanoparticles (NPs) and g-C₃N₄ facilitates directional charge transfer, thereby amplifying photodegradation efficiency. Mechanistic studies confirm that the primary reactive oxygen species (ROS)—·OH, superoxide radicals (·O₂⁻), and photogenerated holes (h⁺)—act synergistically to drive pollutant degradation (Fig. 11b). As illustrated in Fig. 12, the photocatalytic mechanism involves the following cascade: 1) Charge Separation-Upon visible light absorption (h⁺), electrons (e-) are excited from the valence band (VB) of g-C₃N₄ to its conduction band (CB), leaving h⁺ in the VB (Eq. 1). 2) Interfacial Electron Transfer-The staggered band alignment between g-C₃N₄ and ZnFe₂O₄ promotes e- migration from g-C₃N₄ to ZnFe₂O₄ (Eq. 2), where e- reduce adsorbed O₂ to generate ·O₂⁻. 3) ROS Generation-Concurrently, h⁺ in the VB of g-C₃N₄ oxidize H₂O or surface − OH groups to yield ·OH. ·O₂⁻ undergoes protonation to form hydroperoxyl radicals (·OOH), which further react with e- to produce H₂O₂ (Eq. 3). 4) Fenton-like Activation—H₂O₂ is catalytically decomposed via Fe2⁺/Fe3⁺ redox cycles in ZnFe₂O₄, releasing additional ·OH (Eq. 4). 5) Pollutant Mineralization-The synergistic action of ·OH, ·O₂⁻, and h⁺ degrades methylene blue (MB) through demethylation, aromatic ring cleavage, and ultimate mineralization into CO₂, H₂O, and inorganic ions (NH₄⁺/NO₃⁻) (Eq. 5)12,49.

Visible light absorption, charge transfer and schematic diagram of degraded methylene blue by ZnFe2O4@g-C3N4.
The composite’s mesoporous architecture enhances MB adsorption, concentrating dye molecules near ROS-active sites, while the heterojunction’s optimized charge separation minimizes e--h⁺ recombination. This dual functionality—adsorption enrichment and ROS amplification—ensures rapid degradation kinetics and complete pollutant mineralization. Together, these attributes position the ZnFe₂O₄@g-C₃N₄ heterojunction as a sustainable, high-performance photocatalyst for industrial wastewater remediation, with scalability and magnetic recoverability further bolstering its practical viability.
Conclusion
In this study, a ZnFe₂O₄@g-C₃N₄ heterojunction photocatalyst was synthesized via a hydrothermal method. By virtue of its porous multilayer heterojunction interface design, the catalyst achieves efficient photogenerated charge separation and broad-spectrum light absorption. Coupled with a dual-path degradation mechanism—combining photocatalysis and Fenton-like processes (with hydroxyl radicals (·OH) as the dominant active species—the material demonstrated exceptional degradation performance, achieving a 99.99% removal efficiency for methylene blue within 40 min. The reaction rate constant surpassed those of most reported catalysts. Notably, the intrinsic ferromagnetism of ZnFe₂O₄ enabled facile magnetic recovery of the catalyst (retaining > 95% efficiency after 10 cycles). The composite exhibits significant advantages, including cost-effectiveness, facile synthesis, and exceptional stability. Its dual-mechanism synergy (photocatalytic and Fenton-like pathways) highlights its potential for industrial wastewater treatment and environmental remediation, particularly in the degradation of organic pollutants. Furthermore, the heterojunction design strategy proposed here offers a universal framework for developing other visible-light-driven photocatalysts. While further validation is required to assess its efficacy toward complex pollutant systems and complete mineralization pathways, this work provides an innovative and sustainable solution for advanced environmental governance.
Data availability
The datasets used and analysed during the current study available from the corresponding author on reasonable request.
References
Jiang, X. et al. A novel direct Z-scheme heterojunction BiFeO3/ZnFe2O4 photocatalyst for enhanced photocatalyst degradation activity under visible light irradiation. J. Alloy. Compd. 912, 165185 (2022).
Ge, Y. et al. Fabrication and magnetic transformation from paramagnetic to ferrimagnetic of ZnFe2O4 hollow spheres. Trans. Nonferrous Met. Soc. China 29, 1503–1509 (2019).
Wang, M. et al. A facile hydrothermal deposition of ZnFe2O4 nanoparticles on TiO2 nanotube arrays for enhanced visible light photocatalytic activity. J. Mater. Chem. A. 1, 12082–12087 (2013).
Zhao, W. et al. Enhanced photocatalytic and fenton-like performance of CuOx-decorated ZnFe2O4. ACS Appl. Mater. Interfaces. 9, 41927–41936 (2017).
Zhang, Z. L., Wan, M. & Mao, Y. L. Enhanced photovoltaic effect of TiO2-based composite ZnFe2O4/TiO2. J. Photochem. Photobiol., A 233, 15–19 (2012).
Wang, X. et al. Fabrication of a magnetically separable Cu2ZnSnS4/ZnFe2O4 p-n heterostructured nano-photocatalyst for synergistic enhancement of photocatalytic activity combining with photo-Fenton reaction. Appl. Surf. Sci. 479, 86–95 (2019).
Mady, A. H. et al. Facile microwave-assisted green synthesis of Ag-ZnFe2O4@rGO nanocomposites for efficient removal of organic dyes under UV- and visible-light irradiation. J. Phys. Chem. Solids 75, 441–446 (2014).
Yao, Y. et al. Magnetic ZnFe2O4−C3N4 hybrid for photocatalytic degradation of aqueous organic pollutants by visible light. Ind. Eng. Chem. Res. 53, 17294–17302 (2014).
Yang, D. et al. Preparation of 0D/2D ZnFe2O4/Fe-doped g-C3N4 hybrid photocatalysts for visible light N2 fixation. J. Alloy. Compd. 869, 158809 (2021).
Wang, J. & Zhang, W. Oxidative degradation of methylene blue by Ag2O@g-C3N4 photocatalysts under visible light. Toxicol. Environ. Chem. 105, 60–74 (2023).
Wang, J. Construction of ternary heterostructured Ag/Ag2O@ZnO@g-C3N4 nanocomposite as an widened visible light photocatalyst for the organic oxidation. J. Phys. & Chem. Solids. 180, 111389 (2023).
Yang, N. et al. Ternary composite of g-C3N4/ZnFe2O4/Fe2O3: Hydrothermal synthesis and enhanced photocatalytic performance. ChemistrySelect 4, 7308–7316 (2019).
Borthakur, S. & Saikia, L. ZnFe2O4@g-C3N4 nanocomposites: An efficient catalyst for Fenton-like photodegradation of environmentally pollutant Rhodamine B. J. Environ. Chem. Eng. 7, 103035 (2019).
Meng, Y. et al. Construction of g-C3N4/ZIF-67 photocatalyst with enhanced photocatalytic CO2 reduction activity. Mater. Sci. Semicond. Process. 95, 35–41 (2019).
Lu, T. et al. Photocatalysis-self-Fenton system over edge covalently modified g-C3N4 with high mineralization of persistent organic pollutants. Environ. ResearchVolume. 222, 115361 (2023).
Tang, J. et al. Preparation of floating porous g-C3N4 photocatalyst via a facile one-pot method for efficient photocatalytic elimination of tetracycline under visible light irradiation. Chem. Eng. J. 430, 132669 (2022).
Behera, A. et al. Construction of isoenergetic band alignment between CdS QDs and CaFe2O4@ZnFe2O4 heterojunction: A Promising ternary hybrid toward norfloxacin degradation and H2 energy production. J. Phys. Chem. C. 123, 17112–17126 (2019).
Behera, A. et al. Facile synthesis of ZnFe2O4 photocatalysts for decolourization of organic dyes under solar irradiation. Beilstein J. Nanotechnol. 9, 436–446 (2018).
Ghobadifard, M., Farhadi, S. & Mohebbi, S. Catalytic performance of ZnFe2O4 nanoparticles prepared from the [ZnFe2O(CH3COO)6(H2O)3]·2H2O complex under microwave irradiation. Res. Chem. Intermed. 45, 379–400 (2019).
Ghobadifard, M., Farhadi, S. & Mohebbi, S. Sonocatalytic performance of magnetic flower-like CoFe2O4 nanoparticles prepared from a heterometallic oxo-centered trinuclear complex under microwave irradiation. Polyhedron 155, 66–76 (2018).
Ghobadifard, M., Radovanovic, P. & Mohebbi, S. Novel CoFe2O4/CuBi2O4 heterojunction p-n semiconductor as visible-light-driven nanophotocatalyst for C(OH)-H bond activation. Appl. Organomet. Chem. 36, e6612 (2022).
Shi, L. et al. Flower-like Ni(OH)2 hybridized g-C3N4 for high-performance supercapacitor electrode material. Mater. Lett. 145, 150–153 (2015).
Zhang, X. et al. Dual-loading of Fe3O4 and Pd nanoparticles on g-C3N4 nanosheets toward a magnetic nanoplatform with enhanced peroxidase-like activity for loading various enzymes for visual detection of small molecules. Anal. Chem. 95, 5024–5033 (2023).
Pattnaik, S. P., Behera, A., Acharya, R. & Parida, K. Green exfoliation of graphitic carbon nitride towards decolourization of congo-red under solar irradiation. J. Environ. Chem. Eng. 7, 103456 (2019).
Zolfi, F., Ghobadifard, M. & Mohebbi, S. High proficiency Ag/β-Ag2WO4/V3O4/g-C3N4 heterojunction photocatalyst for the actuation of C(OH)-H bond. New J. Chem. 48, 7430–7438 (2024).
Behera, A., Babu, P. & Parida, K. Growth of macroporous TiO2 on B-doped g-C3N4 nanosheets: a Z-scheme photocatalyst for H2O2 production and phenol oxidation under visible light. Inorg. Chem. Front. 8, 1489 (2021).
Zhong, Q. et al. Preparation of heterostructure g-C3N4/ZnO nanorods for high photocatalytic activity on different pollutants (MB, RhB, Cr(VI) and eosin). Ceram. Int. 46, 12192–12199 (2020).
Vijayan, M. et al. Energetic two-dimensional g-C3N4 nanosheets combined with ZnO nanoparticles as effectual catalyst for degradation of MB dye under UV–Visible-light Irradiation. J Mater Sci: Mater Electron. 33, 24340–24353 (2022).
Gao, M. et al. Carbon microspheres work as an electron bridge for degrading high concentration MB in CoFe2O4@carbon microsphere/g-C3N4 with a hierarchical sandwich-structure. Appl. Surf. Sci. 507, 145167 (2020).
Sun, S. et al. Construction of g/C3N4-ZnO composites with enhanced visible-light photocatalytic activity for degradation of amoxicillin. Korean J. Chem. Eng. 39, 3377–3388 (2022).
Veerakumar, P. et al. Nickel nanoparticle-decorated porous carbons for highly active catalytic reduction of organic dyes and sensitive detection of Hg(II) Ions. ACS Appl. Mater. Interfaces. 7, 24810–24821 (2015).
Xie, Y. et al. Highly regenerable mussel-inspired Fe3O4@polydopamine-ag core-shell microspheres as catalyst and adsorbent for methylene blue removal. ACS Appl. Mater. Interfaces. 6, 8845–8852 (2014).
Li, X., Wu, K., Ye, Y. & Wei, X. Controllable synthesis of Ni nanotube arrays and their structure dependent catalytic activity toward dye degradation. CrystEngComm 16, 4406–4413 (2014).
Mirmehdi, H. S. et al. Application of palladium nanoparticle-decorated Artemisia abrotanum extract modified graphene oxide for highly active catalytic reduction of methylene blue, methyl orange and rhodamine B. Appl. Organomet. Chem. 33, e5123 (2019).
Surabhi, K. et al. Synthesis of boron doped C3N4/NiFe2O4 nanocomposite: An enhanced visible light photocatalyst for the degradation of methylene blue. Results Physics. 12, 1238–1244 (2019).
Chen, H. et al. Inorganic-organic hybrid NiO-g-C3N4 photocatalyst for efficient methylene blue degradation using visible light. RSC Adv. 4, 22491–22496 (2014).
Zhang, S. et al. In-situ fabrication of g-C3N4/ZnO nanocomposites for photocatalytic degradation of methylene blue:synthesis procedure does matter. Nanomaterials 9, 215 (2019).
Vadivel, S. et al. Facile synthesis of novel CaFe2O4/g-C3N4 nanocomposites for degradation of methylene blue under visible-light irradiation. J. Colloid Interface Sci. 480, 126–136 (2016).
Fang, Q. et al. 0D/2D Z-scheme heterojunctions of g-C3N4 quantum dots/ZnO nanosheets as a highly efficient visible-light photocatalyst. Adv. Powder Technol. 30, 1576–1583 (2019).
Guo, X., Wang, K., Li, D. & Qin, J. Heterogeneous photo-Fenton processes using graphite carbon coating hollow CuFe2O4 spheres for the degradation of methylene blue. Appl. Surf. Sci. 420, 792–801 (2017).
Fardin, G. P. et al. Synthesis of Fe3O4/SiO2/TiO2-Ag photo-catalytic nano-structures with an effective silica shell for degradation of methylene blue. J. Inorg. Organomet. Polym Mater. 30, 3740–3749 (2020).
Hong, M. T. et al. Synthesis and characterization of N-doped graphene oxide quantum dots/Fe-BDC composite for methylene blue decomposition.Chemical Engineering Communications. 211, 428-441 (2024).
Nuengmatcha, P. et al. Sonocatalytic performance of ZnO/graphene/TiO2 nanocomposite for degradation of dye pollutants (methylene blue, texbrite BAC-L, texbrite BBU-L and texbrite NFW-L) under ultrasonic irradiation. Dyes Pigm. 134, 487–497 (2016).
Qin, J. et al. ZnO microspheres-reduced graphene oxide nanocomposite for photocatalytic degradation of methylene blue dye. Appl. Surface Sci. 392(196), 203 (2017).
Huo, Q. et al. ZnO-rich CdS-ZIF-8 catalyst for enhanced visible-light photocatalytic degradation of methylene blue. Res Chem Intermed. 44, 2347–2364 (2018).
Ge, H. et al. Synthesis of citric acid functionalized magnetic graphene oxide coated corn straw for methylene blue adsorption. Biores. Technol. 221, 419–429 (2016).
Fardin, G. P., Saeideh, D. & Fatemeh, B. Synthesis of Fe3O4/SiO2/TiO2-Ag photo-catalytic nano-structures with an efective silica shell for degradation of methylene blue. J. Inorg. Organomet. Polym Mater. 30, 3740–3749 (2020).
Li, Y. et al. Solvent-free synthesis of magnetic biochar and activated carbon through ball-mill extrusion with Fe3O4 nanoparticles for enhancing adsorption of methylene blue. Sci. Total Environ. 722, 137972 (2020).
Nguyen, C. H., Fu, C. C. & Juang, R. S. Degradation of methylene blue and methyl orange by palladium-doped TiO2 photocatalysis for water reuse: Efficiency and degradation pathways. J. Clean. Prod. 202, 413–427 (2018).
Funding
This project was founded by the Liaoning Natural Science Foundation [Nos. 2020-Ms-306], Innovation and Entrepreneurship Training Program of Shenyang University [202411035016].
Author information
Authors and Affiliations
Contributions
Leyan LI-article writing, chart output, grammar correction;Jianhua Wang-material synthesis, article writing, chart output, grammar correction, etc. Huihui FANG-Material preparation, degradation experiments, grammar correction, etc. Jianhua Wang as corresponding authors: Conceptualization, Methodology, Software, Data curation, Writing- Original draft preparation, Validation, Writing- Reviewing and Editing, Funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no known competing for financial interest or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, L., Jianhua, W. & Fang, H. Fabrication of ZnFe2O4@g-C3N4 for enhanced photo-fenton effect and visible light-driven organic dye degradation. Sci Rep 15, 21707 (2025). https://doi.org/10.1038/s41598-025-05096-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-05096-9
