
Abstract
Colorectal cancer (CRC) is the third most common cancer and the second leading cause of cancer-related deaths worldwide. Increasing evidence has shown that ferroptosis plays a significant regulatory role in the occurrence and progression of cancer. However, new biomarkers associated with ferroptosis and new ferroptosis-related subtypes in CRC remain to be identified. We collected a colon adenocarcinoma (COAD) dataset from the cancer genome atlas (TCGA) database in UCSC Xena. Ferroptosis-related genes (FRGs) were extracted from the dataset, and we conducted gene expression profiling interactive analysis (GEPIA), cBioPortal analysis, and immune cell infiltration analysis. Additionally, we used the “ConsensusClusterPlus” R package to identify ferroptosis-related subtypes of CRC. Based on these subtypes, we analyzed their prognostic features, gene co-expression networks, and the ferroptosis landscape. We identified 21 FRGs in COAD. The genes RBMS1, NOX4, FABP4, CYB5R1, CPEB1, and ATM were significantly and positively correlated with immune cell infiltration and poor prognosis. Furthermore, we identified two COAD subtypes related to prognosis and ferroptosis (ferroptosis subtype 1 [FS1] and ferroptosis subtype 2 [FS2]). These subtypes were associated with tumor mutation burden (TMB), mutation status, immunogenic cell death (ICD), and immune checkpoint (ICP) regulatory genes. Finally, we established a ferroptosis landscape of COAD. We identified key ferroptosis-related genes and defined two distinct ferroptosis-associated subtypes in CRC that differ significantly in prognosis and immune infiltration. These findings provide new insights into the interaction between ferroptosis and the tumor immune microenvironment, offering potential biomarkers and therapeutic targets for improved diagnosis and personalized treatment of colorectal cancer.
Similar content being viewed by others
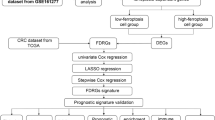


Introduction
Colorectal cancer (CRC) is an aggressive cancer type with a high incidence worldwide, significant morbidity, and poor survival1. Various factors, such as aging, a Western diet, and gene mutations caused by reactive oxygen species (ROS), typically promote the development of CRC2,3.
Although surgery, radiotherapy, chemotherapy, screening, and early diagnosis have rapidly advanced in the treatment of CRC4, few patients respond well or benefit significantly from these diagnostic methods and therapies. Therefore, it is essential to investigate the underlying molecular mechanisms to identify biomarkers for translational research.
Programmed cell death (apoptosis), which involves a series of intracellular molecular steps, is a physiological process through which the body eliminates unwanted or aberrant cells. Cancer cells often evade this response. In 2012, Dr. Brent R. Stockwell identified a novel mode of intracellular iron-related cell death called ferroptosis5. The major features of ferroptosis include an iron-dependent mechanism by which ROS triggers lipid peroxidation in the cell. As a crucial form of cell death, ferroptosis is implicated in the pathogenesis of diseases such as cancer, cardiovascular disease, and neurological disorders. Ferroptosis is distinct from traditional cell death pathways such as apoptosis, necroptosis, and pyroptosis in both its morphological and biochemical mechanisms6. During the reprogramming of cancer metabolism, the ferroptosis pathway can be altered, thereby affecting downstream biological processes7. Given that the gastrointestinal tract is a primary site for lipid and iron metabolism, it provides a permissive environment for ferroptosis, influencing both innate and adaptive immunity by altering immune cell function and promoting DAMP-mediated immune responses through the death of non-immune cells8. Therefore, understanding the mechanisms of ferroptosis is critical for overcoming resistance to tumor therapies. Inducing ferroptosis in CRC cells by targeting key regulators such as glutathione (GSH), GPX4, and SLC7A11 is being explored as a promising therapeutic strategy. For instance, cetuximab has been shown to modulate ferroptosis to enhance chemotherapy efficacy9. Predicting CRC carcinogenesis using tumor-specific antigens has also attracted increasing interest among clinicians10.
In the present study, we employed bioinformatics methods to identify differentially expressed genes (DEGs) in CRC patients compared to healthy controls. From these DEGs, we identified ferroptosis-associated genes, which were subsequently used to determine ferroptosis subtypes in CRC. We further analyzed the cellular and molecular characteristics of these subtypes and constructed the ferroptosis landscape of colon adenocarcinoma (COAD). These findings are important for understanding the underlying mechanisms and guiding CRC therapy.
Materials and methods
Data acquisition and pretreatments
The COAD dataset from the NCI Genomic Data Commons (GDC) data portal. The Cancer Genome Atlas (TCGA) Colon Cancer (n = 512) was downloaded via the UCSC Xena database (https://xena.ucsc.edu/). The data types “Count” and “FPKM” were selected, and “Primary Solid Tumor” (01A) samples were extracted and converted to the TPM format. “Masked Somatic Mutation” data were selected as the somatic mutation data for COAD patients and were preprocessed using VarScan software. Somatic mutation data were visualized using the maftools R package. In addition, patient clinical data, including age, TNM stage, survival time, and survival state, were downloaded. Patients lacking clinical information were excluded. In total, 412 patients with survival data and 331 with other clinical information were retained. Gene expression data (n = 585) from GSE39582 and clinical information, including survival time and survival state (n = 579), were downloaded from the GEO database. The samples were derived from Homo sapiens, and the chip platform was used was GPL570. Finally, 579 tumor samples were retained for this study, and the microarray data were normalized using the limma package in R.
We also extracted ferroptosis-related genes (FRGs) from FERDB (www.zhounan.org/ferrdb/), including driver genes, suppressor genes, and markers, and identified a total of 424 FRGs.
Identification of tumor antigens
GEPIA and cBioPortal analysis
RNA-Seq raw data downloaded from the TCGA database were recalculated via UCSC Xena to prevent data imbalance and inefficiencies in differential analysis. Differentially expressed genes were identified using Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/). DEGs with |log2FC| values > 1 and q values < 0.05 were identified using ANOVA. Overall survival (OS) and progression-free survival (PFS) were assessed using the Kaplan–Meier method with median cutoff values and log-rank tests. P values < 0.05 were considered statistically significant.
The cBio Cancer Genomics Portal (http://www.cbioportal.org) was used to integrate RNA-seq data from databases such as TCGA, and gene alterations in COAD were compared. Microsatellite instability and tumor mutation load data were extracted from TCGA-COAD samples. A P-value < 0.05 was considered statistically significant.
Tumor antigen and immune cell infiltration
We used the Tumor Immunoassay Resource (Timer, https://cistrome.shinyapps.io/timer/) to analyze and visualize the relationship between the abundance of tumor immune infiltrating cells (TIICs) and potential tumor antigens. Analysis modules included gene expression, somatic mutation, clinical outcome, and somatic copy number alterations. Spearman correlation analysis was used, and purity-adjusted correlations were calculated. P values < 0.05 were considered statistically significant.
Additionally, the MCPcounter method was used to estimate the infiltration levels of tumor-infiltrating immune and stromal cells from gene expression data. MCPcounter provided abundance estimates for eight immune cell populations: CD4 + T cells, CD8 + T cells, natural killer (NK) cells, B lymphocytes, monocytes, dendritic cells, neutrophils, and cytotoxic lymphocytes (CD8 + T cells and cytotoxic innate lymphoid NK cells). Abundance estimates for two stromal cell types (fibroblasts and endothelial cells) were also generated. Spearman correlation analysis was performed between the MCPcounter-derived cell abundance and antigen gene expression; with P value < 0.05 considered significant.
Discovery and verification of ferroptosis subtypes
The “ConsensusClusterPlus” R package was used to cluster ferroptosis-related genes based on expression patterns and to establish a consensus matrix identifying ferroptosis subtypes and gene modules. We first applied the k-means clustering algorithm using the Euclidean distance. The optimal number of clusters was determined by evaluating the area under the cumulative distribution function (CDF) curve11. Partitioning was performed using a median algorithm via a “1-Pearson correlation” distance measure; 1,000 replicates were conducted, and each resampling was 80% of the patients in the cohort. We then analyzed the association with the “Survminer” R package. Differentially expressed ferroptosis-related genes (DE-FRGs) among subtypes were identified via ANOVA with Benjamini–Hochberg adjusted P-values (adjusted P < 0.05)12. Pearson correlation analysis was used to evaluate co-expression patterns of FRGs in CRC. Principal component analysis (PCA) was performed using the “psych” R package to visualize subtype distribution based on DE-FRG expression profiles. The cluster range was set from 2 to 9, and the optimal number was determined using the consensus matrix and the cumulative distribution function. Ferroptosis subtypes were validated in GSE39582 using the same approach.
Prognostic evaluation of ferroptosis subtypes
The prognostic significance of ferroptosis subtypes was evaluated using log-rank tests. ANOVA was used to analyze correlations between ferroptosis subtypes and molecular/cellular features. The most frequently mutated genes were identified using the chi-squared test. Single-sample Gene Set Enrichment Analysis (ssGSEA) of the Gene Set Variation Analysis (GSVA) package was applied to calculate the immune enrichment scores for each sample.
Gene co-expression network
The “WGCNA” R package was used to construct immune-related gene co-expression networks. The soft threshold was determined via the pickSoftThreshold function. The optimal soft threshold was determined to be six, and then the scale-free network was established based on the soft threshold. Subsequently, the topology matrix was established, and hierarchical clustering was performed to calculate eigengenes. Using the Eigengenes module, the correlation between modules was constructed, and hierarchical clustering was performed. Functional enrichment analyses were performed via the Metascape database (www.metascape.org/). This included the Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO), REACTOME Gene Sets, Canonical Pathways, CORUM, and Wiki Pathways. A P value < 0.05 was considered significantly enriched.
Landscape construction of ferroptosis in the tumor microenvironment
A graph-learning-based dimensionality reduction analysis and the dimensionality reduction function of the Monocle package with a Gaussian distribution were applied to visualize the distribution of the ferroptosis subtypes in single patients. The maximum number of components was set to four, and the discriminant dimension reduction means of “DDRtree” was applied. Finally, the cell track was applied to visualize the ferroptosis landscape using the functional diagram of the color-coded ferroptosis subtypes.
Statistical analysis
All statistical analyses were conducted using R software (version 4.1.1). The differences between the two groups were analyzed via the Wilcoxon signed-rank test. Differences between the two groups were assessed using the Kruskal–Wallis test. The Spearman correlation method was applied for correlation analyses. A P < 0.05 was the statistically significant threshold.
Results
Identification of potential ferroptosis-related antigens in COAD
To support the development of a potential COAD mRNA vaccine13, we first screened 5352 DEGs, identifying 2673 overexpressed genes that may encode tumor-associated antigens (Fig. 1A). We also screened 18,275 mutant genes by analyzing mutated gene fragments and mutation counts per sample (Fig. 1B,D). Mutation analysis indicated that APC and TP53 were the most frequently mutated genes, based on both mutant genome fragments and mutation counts (Fig. 1C,E). Regardless of mutation frequency, genes such as APC, TP53, TTN, KRAS, and RBFOX1 exhibited significant mutation rates in COAD. However, due to the long sequence and low specificity of TTN, other gene mutations may be more relevant in COAD. Based on the mutant and overexpressed gene results, we identified 413 cancer-related genes with frequent mutations and upregulation that may serve as potential tumor antigens.

Identification of potential tumor antigens of COAD. Identification of potential tumor-associated antigens in COAD, showing the chromosomal distribution of upregulated genes in COAD (A). The distribution relationships between different mutation count groups and subtypes of colorectal adenocarcinoma patients (B). The distribution and statistical significance of chromosomal arm copy number alterations (CNAs) within different mutation count groups (C). The distribution relationships between different fraction genomes altered groups and subtypes of colorectal adenocarcinoma patients (D). The distribution and statistical significance of CNAs within different fraction genome altered groups (E). COAD colon adenocarcinoma, CNAs copy number alterations.
Next, we investigated whether ferroptosis-related genes could serve as mRNA vaccine antigens. After analysis of overall survival and PFS, 21 FRGs were identified from 462 potential antigens in this study. Considering the interaction of tumor antigens with antigen-presenting cells, we further screened the relationship of these potential antigens with antigen-presenting cells (APCs) using the MCPcounter binding TIMER database. The RBMS1, NOX4, FABP4, CYB5R1, CPEB1, and ATM genes were significantly positively correlated with immune and stromal cells (Fig. 2A). This was evidenced by the results of the TIMER database, especially regarding macrophages (Fig. 2B–E). Survival analysis revealed that high expression of these genes was associated with poorer OS and PFS (Fig. 3A–L). Moreover, expression of these genes tended to increase with advancing TNM stage (Fig. S1A–D). These findings suggest that these tumor antigens may be processed by APCs, presented to T cells, and induce immune responses. Furthermore, they may also participate in inducing ferroptosis to eliminate tumor cells.

Identification of APCs-associated tumor antigens. Analysis of association of immune and stromal cells with the RBMS1, NOX4, FABP4, CYB5R1, CPEB1, and ATM genes, calculated by MCP COUNTER (A). Correlation of expression levels of ATM (B), CPEB1 (C), CYB5R1 (D), FABP4 (E), NOX4 (F), and RBMS1 (G) in COAD tumors with tumor purity and infiltration of B cells, macrophages, and dendritic cells. ACPs antigen presenting cells, COAD colon adenocarcinoma.

Identification of tumor antigens relevant to the prognosis of COAD. Kaplan–Meier curves showing OS and PFS stratified by gene expression levels of ATM (A,G), CPEB1 (B,H), CYBR1 (C,I), FABP (D,J), NOX4 (E,K), and RBMS1 (F,L) in COAD patients. OS overall survival, PFS progression-free survival, COAD colon adenocarcinoma.
Identification of potential COAD ferroptosis subtypes
Ferroptosis plays a crucial role in tumor cell death and is closely linked to tumor immunity. Ferroptosis subtypes may reflect the ferroptosis state of tumors and their microenvironments, aiding in the identification of patients eligible for vaccination. Expression profiles of 424 FRGs in the TCGA-COAD cohort were analyzed using consensus clustering. Based on their cumulative distribution function and functional trigonal area, we selected k = 2, where immune-related genes steadily aggregated (Fig. 4A,B), and identified two ferroptosis subtypes (ferroptosis subtypes 1 (FS1) and ferroptosis subtypes 2 (FS2)) (Fig. 4C). GSE39582 data were similarly classified into FS1 and FS2 (Fig. 4D). PCA confirmed distinct distributions between the two subtypes (Fig. 4E). Prognostic analysis showed that FS1 had better survival than FS2 in both the TCGA and GSE39582 cohorts (Fig. 4F,G). In GSE39582, FS2 cases were more frequent in intermediate and advanced stages (Fig. S2A,B). Thus, ferroptosis subtyping predicts prognosis and is reproducible across datasets.

Identification of potential ferroptosis subtypes of COAD. Cumulative distribution function (A) and δ area (B) of genes associated with ferroptosis in TCGA-COAD cohort. TCGA-COAD sample K = 2 clustering heat map (C). GSE39582 Sample K = 2 Cluster heat map (D). The principal component analysis of ferroptosis subtypes in TCGA-COAD (E). Kaplan–Meier curve demonstrates the OS of the COAD ferroptosis subtype in the TCGA cohort (F). Kaplan–Meier curve shows the OS of the COAD ferroptosis subtype in GSE39582 Cohort (G). COAD colon adenocarcinoma, TCGA the cancer genome atlas, OS overall survival.
Relationship between ferroptosis subtypes and mutation state
Somatic mutation rate and tumor mutation burden (TMB) are positively correlated with anti-tumor immunity. Thirty genes, including APC, TP53, TTN, KRAS, SYNE1, PIK3CA, and MUC16, showed subtype-specific mutation patterns (Fig. 5A). Subsequently, we determined the TMB, MSI, and the total number of mutations for each COAD patient using the mutation data set in TCGA-COAD and compared ferroptosis subtypes (Fig. 5B–D). As shown in Fig. 5C, the total number of TMBs in the FS2 subtype was higher than in the FS1 subtype, and the statistical difference between the two groups was striking (P < 0.05). There was no remarkable difference in the total mutation number. In addition, the MSI differences between the two ferroptosis subgroups were not significant (Fig. 5D). The above results suggest that TMB may be a potential indicator for different ferroptosis subtypes and mutation characteristics.

Relationship between ferroptosis subtypes and TMB and mutations. COAD waterfall plot shows characteristics of mutated genes from different ferroptosis subtypes (FS1 and FS2) (A). Comparation of Mutation count (B), TMB (C), and MSI (D) among different ferroptosis subtypes. *P < 0.05, ns not significant. COAD colon adenocarcinoma, TMB tumor mutation burden, MSI microsatellite instability, FS1 ferroptosis subtypes 1, FS2 ferroptosis subtypes 2.
The relationship between COAD ferroptosis subtypes and immunomodulators
Previous studies have demonstrated that immune checkpoints (ICPs) (such as PD-L1 and TIM-3) and immunogenic cell death (ICD) modulators (such as CALR and HMGB1) perform vital roles in the regulation of the anti-tumor immunity of the host. Moreover, ferroptosis has an important relationship with immune regulation. Therefore, we evaluated the differential expression of both ICP and ICD modulators in two ferroptosis subtypes. We tested 25 ICD genes in the TCGA-COAD cohort and found 14 ICD genes differing between the two subtypes (Fig. 6A). In addition, 23 ICD genes in the GSE39582 cohort were tested, and 18 of these were distinct between the two subtypes (Fig. 6B). We observed that the differentially expressed ICD genes in different subtypes exhibited similar expression trends. For example, genes such as ANXA1, CXCL10, and PANX1 were decreased in the FS1 subtype (Fig. 6A,B). Although there were many molecules whose expression did not show significant statistical differences, most ICD gene expression trends were consistent between the two data sets. In addition, 46 ICP genes in the TCGA-COAD cohort were identified, 33 of which were differentially expressed in the two subtypes (Fig. 6C). Additionally, 43 ICP genes were tested in the GSE39582 Cohort, and 24 of these were differentially expressed in the two subtypes (Fig. 6D); ICP gene expression trends were consistent across the two cohorts. In summary, the ferroptosis classification reflected the expression of ICD modulators and ICPs and may be applied as a biomarker for immunotherapy.

Relationship between ferroptosis subtypes and ICD and ICP regulatory genes. Difference in ICD modulator expression between the two ferroptosis subtypes, FS1 and FS2, in the TCGA-COAD cohort (A) and the GSE39582 cohort (B). Differences in ICD expression between the two ferroptosis subtypes FS1 and FS2, in the TCGA-COAD cohort (C) and the GSE39582 cohort (D). *P < 0.05, **P < 0.01, ***P < 0.001. ICD immunogenic cell death, ICP immune checkpoint, FS1 ferroptosis subtypes 1, FS2 ferroptosis subtypes 2, TCGA the cancer genome atlas, COAD colon adenocarcinoma.
Cellular and molecular features of ferroptosis subtypes
The immune state of the tumor determines the reaction to mRNA vaccines. Based on the ESTIMATE algorithm, we found greater immunocyte infiltration in FS2 tumor patients in both TCGA-COAD and GSE39582, where the ESTIMATE score and stromal cell score only indicated that FS2 was higher than FS1 in TCGA-COAD (Fig. 7A–F). Subsequently, the immune cell components in the two ferroptosis subtypes were further characterized by calculating the score of 28 signature genes previously reported in the TCGA-COAD and GSE39582 cohorts through ssGSEA. As illustrated in (Fig. S3A,B), the immune cell profiles formed two clusters. The immune cell composition of different subtypes was different. The immune cell enrichment score showed that patients with the FS2 subtype had more immune cell infiltration (Fig. S3C,D). The KM curve showed that among the 22 immune cells, memory CD4 + T cells and activated CD4 + T cells were associated with different prognoses, and both of these showed that the prognosis was better with a higher score (Fig. 7G). When comparing the above two immunocytes with different prognoses, they both showed significant differences in the TCGA-COAD (Fig. 7H) and GSE39582 data sets (Fig. 7I). Hot tumors are typically characterized by high levels of T cell infiltration, elevated PD-L1 expression, and increased TMB, making them more responsive to immune checkpoint inhibitors14. Ferroptosis enhances the efficacy of immune checkpoint inhibitors by remodeling the tumor microenvironment, and targeted delivery of ferroptosis inducers can convert “cold” tumors into “hot” tumors, promoting precision immunotherapy15. Recent studies reveal that ferroptosis-mediated reprogramming of the tumor microenvironment by crosstalk between cancer and non-cancer cells plays a crucial role in tumor progression and therapeutic resistance16. In summary, FS2 may be the immune “hot” phenotype, while FS1 may be the immune “cold” phenotype. We performed GO and KEGG enrichment analyses on the differentially expressed genes between FS1 and FS2. The results revealed that FS2 is significantly enriched in immune-related pathways, including T cell differentiation, cytokine-cytokine receptor interaction, and antigen processing and presentation. In contrast, FS1 is more enriched in metabolism-related pathways. These findings support the classification of FS2 as an immune “hot” phenotype and FS1 as an immune “cold” phenotype. (Fig. S4A,B). Although our bioinformatics analyses suggest that FS1 and FS2 represent immune “cold” and “hot” phenotypes, respectively, further experimental validation (e.g., immunohistochemistry, flow cytometry, functional assays) will be necessary to confirm the immune landscape and biological behaviors of these subtypes.

Cellular and molecular features of ferroptosis subtypes. ESTIAMTE (A), immune (B), and matrix (C) score in the TCGA-COAD data set. ESTIAMTE scores (D), immune scores (E), and matrix (F) scores in the GSE39582 data set. In TCGA-COAD, obvious prognostic differences exist in the high and low scores of effector memory CD4 + T cells and activated CD4 + T cells (G). In TCGA-COAD, the high and low scores of memory CD4 + T cells and activated CD4 + T cells are different (H). In GSE39582, high and low scores of memory CD4 + T cells and activated CD4 + T cells have significant differences (I). TCGA the cancer genome atlas, COAD colon adenocarcinoma, ESTIMATE estimation of STromal and immune cells in MAlignant tumor tissues using expression data.
These findings suggest that the ferroptosis subtype reflects the immune state of COAD and may identify patients appropriate for mRNA inoculation. These mRNA-containing antigens could evoke immune infiltration in patients with low immunocompetence diagnosed with FS2 tumors.
COAD Ferroptosis landscape
The COAD ferroptosis profile was constructed using the ferroptosis gene expression profile from each patient (Fig. 8A). As illustrated in Fig. 8B, the horizontal axis (PCA1) was related to various immune cells and exhibited a significant positive correlation with mononuclear cells, memory B cells, mast cells, and γδ T cells. The vertical axis (PCA2) was positively associated with NK cells, monocytes, MDSC, and γδ T cells and was negatively associated with activated CD4 + T cells and type 2 helper T cells. We also observed different levels of intra-cluster heterogeneity within the same subtype. Based on the locus of the sample population, we further divided all the samples into five states (Fig. 8C). We selected the terminal state for analysis, including 1-, 3-, and 5-year states. Their proportions among the different ferroptosis subtypes are shown in (Fig. 8D). The prognostic analysis demonstrated survival differences in the overall survival curves of the three states. Among these, the prognosis of state 1 was the worst, and the survival curves of states 3 and 5 significantly crossed (Fig. 8E). In summary, the ferroptosis map based on the ferroptosis subtype was unable to precisely identify the ferroptosis status of every patient with COAD and forecast their prognosis. Further verification with more samples and typing verification may be required.

The ferroptosis landscape of COAD. The position and color of each patient in the immune map correspond to the above-identified ferroptosis subtypes, representing the overall characteristics of FRME (A). Interrelation between the immune cell module and PCA1/2 (B). COAD patients are reclassified according to their ___location, and they are divided into 5 states (C). The proportion of ferroptosis classification of state patients at the endpoint (D). Prognostic curves of patients with different states (E). It can be found that the prognosis of state 1 patients is worse, while the survival curves of other state groups have no significant difference. COAD colon adenocarcinoma.
Determination of the ferroptosis center gene and ferroptosis gene co-expression module of COAD
To identify key genes, a weighted correlation network analysis (WGCNA) of immune-related genes was built in the scale-free network; the soft threshold was determined to be three (Fig. 9A). The gene matrix was transformed into an adjacent matrix and then into a topology matrix. A minimum module size of 20 genes was set. The characteristic genes of every module were added, and similar modules were combined. Finally, three modules were obtained, where the gray module contained unassigned genes (Fig. 9B,C). When comparing the eigengene scores of the modules, we found that the PS1 score was significantly lower than the PS2 score, except in the gray module (Fig. 9D). To identify independent prognostic modules, we then performed a multivariate Cox analysis of the eigengene scores on the modules. However, multivariate Cox results showed that not all the modules were independent prognostic factors (Fig. 9E). Subsequently, we extracted the blue module (MEblue) that had the highest correlation with the ferroptosis subtype and conducted functional enrichment of genes in the module. The GO enrichment indicated that most of the blue module genes were enriched in the category of immunity. Pathway enrichment was mainly involved in Cytokine-Cytokine Receptor Interaction and Antigen Processing and Presentation (Fig. S4A,B). Among these, the hub genes in the blue module included IL-32, FCGR 2b, LYZ, FGFR4, IL-10RA, and STAT1. Therefore, these hub genes could serve as a biomarker to forecast the prognosis of COAD patients and identify patients for mRNA vaccination.

Identification of COAD core ferroptosis gene. The optimal soft threshold value of WGCNA is determined and the selected soft threshold value is 3 (A). 3 modules were obtained by WGCNA (B,C). Comparison of scores of ferroptosis subtypes in different modules (D). Multivariate Cox analysis of different module scores (E). COAD colon adenocarcinoma, WGCNA weighted gene co-expression network analysis.
Discussion
Ferroptosis is a novel, iron-dependent type of programmed cell death that differs from necrosis, autophagy, and apoptosis. The main mechanism of ferroptosis is the induction of cell death through lipid peroxidation. This results from the catalysis of unsaturated fatty acids, which are highly expressed on the cell membrane, in the presence of divalent iron or ester oxygenase. Moreover, studies have shown that regulation of the core antioxidant system (glutathione system), the GPX4 is decreased17.
Anna Konstorum et al. employed a systems-level approach using five key variables—ACSL4, SCD1, ferroportin, transferrin receptor, and p53—to classify ovarian cancer into high and low ferroptosis sensitivity groups, demonstrating that such modeling can effectively predict cancer susceptibility to ferroptosis12. Using weighted gene correlation network analysis (WGCNA), Zhang et al. identified 21 ferroptosis-related genes (FRGs) associated with amyotrophic lateral sclerosis (ALS), revealing that higher expression levels of CHMP5 and SLC38A1 in whole blood are linked to shorter patient survival18. Ferroptosis risk signatures may predict CRC prognosis and the sensitivity of patients to targeted therapy. However, new biomarkers associated with ferroptosis and new ferroptosis-related subtypes of CRC remain to be identified.
Dai et al. analyzed the GSE130970 dataset and classified MAFLD patients into two distinct subtypes, one of which showed upregulation of eight ferroptosis-related genes, with significant differences between the subtypes in hepatic steatosis grade (P < 0.05), NAS (P < 0.005), and immune status heterogeneity19. In our study, we identified 21 FRGs in COAD. Among these genes, RBMS1, NOX4, FABP4, CYB5R1, CPEB1, and ATM were significantly positively correlated with immune cell infiltration and poor prognosis. In xenograft mouse models, NOX4 plays a critical role in promoting ferroptosis-associated tumor progression, as its inhibition blocks cell death, indicating that NOX4 contributes to poor prognosis and may serve as a potential therapeutic target under cystine-depleted conditions20. Both in vivo and in vitro experiments have demonstrated that RBMS1 promotes the translation of prion protein, leading to ferroptosis resistance in tumor cells, while RBMS1 inhibition induces ferroptosis in colorectal cancer cells and restores their sensitivity to oxaliplatin21. Furthermore, we identified two prognosis- and ferroptosis-related COAD subtypes (FS1 and FS2). We also showed that different ferroptosis subtypes were associated with TMB, mutation status, and ICD and ICP regulatory genes. Finally, we identified the cellular and molecular characteristics of the ferroptosis subtypes in different cohorts, thereby establishing a ferroptosis landscape for COAD.
RNA-binding motif, single-stranded-interacting protein 1 (RBMS1) is a c-Myc-enhancer binding factor that is typically expressed in latency III of EBV and is related to the lytic cycle22. RBMS1 plays an important role in colon cancer metastasis23, prostate cancer development24, gastric cancer prognosis25, lung cancer ferroptosis, and breast cancer immunotherapy26. In addition, RBMS1 (FS2 Subtype) was highly expressed in oxaliplatin-resistant colorectal cancer cells, and its depletion in HCT116 L-OHP cells restored ferroptosis-induced cell death and enhanced the therapeutic efficacy of oxaliplatin in vivo21. NOX4 is overexpressed in CRC and is associated with poor prognosis and tumor progression27. Meanwhile, NOX4 (FS2 Subtype) suppresses ferroptosis by modulating G6PD activity and regulating the pentose phosphate pathway (PPP). Targeting NOX4 or limiting PPP flux through G6PD or GLUT inhibitors may reveal a metabolic vulnerability for therapy28. Additionally, NOX4 co-expresses with ANGPTL4 to promote metastasis29 and is involved in angiogenesis, epithelial-mesenchymal transition (EMT), and Notch signaling30. The fatty acid-binding protein 4 (FABP4) was shown to be overexpressed in CRC and related to cancer stage and metastasis. Mechanistically, FABP4 may enhance CRC progression through EMT31. The roles of FABP4, CYB5R1, CPEB1, and ATM in CRC-related ferroptosis remain unstudied and warrant further investigation. CYB5R1 was also shown to be correlated with EMT and poor survival in CRC32. Furthermore, the function of CYB5R1 in CRC ferroptosis has not been studied. FABP4 inhibits ferroptosis by promoting lipid droplet formation and supporting immunosuppressive macrophage polarization, thereby facilitating tumor immune evasion, while CYB5R1 cooperates with enzymes like POR to enhance lipid peroxidation and increase ferroptosis sensitivity in cancer cells33. Additionally, NOX4 regulates reactive oxygen species production to promote ferroptosis and modulate PD-L1 expression, influencing immune escape, whereas CPEB1 promotes ferroptosis through activation of the ATF4/CHAC1 pathway and modulation of NRF2-mediated antioxidant responses, collectively highlighting their complex roles in ferroptosis regulation and tumor progression34. ATM in the DNA damage response pathway is important in the treatment of many different types of cancer35. The function of the ATM in CRC ferroptosis has not been studied. Therefore, the function of these molecules in CRC ferroptosis should be further investigated.
Using our bioinformatic tools, we further analyzed ferroptosis subtypes in CRC. Other studies have analyzed ferroptosis subtypes in cancer. Zhu36 et al. discovered that ferroptosis-related genes associated with overall survival and prognosis in liver cancer are regulated by non-coding RNAs through m1a, m5c, and m6a epigenetic modifications. Besides, silencing PRDX1 in HEPG2 cells could lead to increased accumulation of ferrous iron and lipid peroxidation in hepatocellular carcinoma37. Yang 38 et al. used LASSO COX regression to identify a ferroptosis-related CRC prognosis model, and they characterized five groups associated with forecasting prognosis in CRC. Yue39 et al. applied consensus clustering to obtain four ferroptosis-related CRC subtypes, which highlighted notable differences in drug sensitivity among the subtypes, showing variable responses to gemcitabine and bortezomib (P < 0.0001), with the C3 subtype specifically exhibiting resistance to erlotinib. Moreover, we used different tools such as the “ConsensusClusterPlus” R package and obtained different results. We identified two CRC ferroptosis subtypes, which we designated FS1 and FS2. The two ferroptosis subtypes were associated with CRC prognosis in both the training and validation datasets. We also showed that the different ferroptosis subtypes had different mutation characteristics. Moreover, our ferroptosis classification indicated that the expression levels of the ICD modulator and ICPs may be used as biomarkers to indicate appropriateness for mRNA vaccination. The ferroptosis subtype reflected the immune state of COAD and may be used to identify patients who would benefit from mRNA inoculation. Finally, we established a ferroptosis landscape for COAD.
Our study had several limitations. First, a clinical cohort is needed to validate the molecular stratification of CRC, and further verification with larger, independent sample sets and clinical typing is warranted. The subtypes FS1 and FS2 were primarily defined based on computational analysis using the TCGA-COAD dataset and were subsequently validated in the independent GSE39582 cohort. However, we acknowledge that no direct biological validation (such as IHC staining, qPCR, or ferroptosis-specific functional assays) was performed in this study. Second, our method of CRC subtyping should be compared to existing classification approaches to assess its robustness and clinical relevance. Third, although we identified survival-associated FRGs, the mechanistic links between these genes and the molecular subtypes of CRC remain incomplete; additional analyses, such as correlation studies and further bioinformatics validation, are needed to bridge this gap. Moreover, potential batch effects and platform biases may have influenced the results, which should be considered when interpreting the findings. Finally, the lack of independent experimental validation limits the translational strength of our conclusions, and this will be addressed in future studies.
Conclusion
In this study, we comprehensively explored the role of ferroptosis in CRC through integrative bioinformatic analyses. We identified 21 FRGs in COAD, including RBMS1, NOX4, FABP4, CYB5R1, CPEB1, and ATM, which were closely associated with immune cell infiltration and poor prognosis. Based on the expression profiles of FRGs, we classified CRC patients into two distinct ferroptosis-related subtypes, FS1 and FS2. These subtypes exhibited significant differences in prognosis, TMB, ICP expression, ICD modulators, and immune microenvironment characteristics. Furthermore, we established a ferroptosis landscape of CRC and identified key ferroptosis-associated gene modules that may serve as potential prognostic biomarkers and therapeutic targets.
Our findings provide new insights into the interplay between ferroptosis and tumor immunity and suggest a ferroptosis-based classification system that may guide personalized immunotherapy for CRC. Nevertheless, future experimental validation and clinical cohort studies are essential to confirm the biological relevance and translational value of these findings.
Data availability
The data utilized in this study are publicly available from the GDC and GEO repository, which can be accessed at https://docs.gdc.cancer.gov/Data/Release_Notes/Data_Release_Notes/#data-release-400 and https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE39582amp;format=file.
References
Labianca, R. et al. Colon cancer. Crit. Rev. Oncol. Hematol. 74, 106–133. https://doi.org/10.1016/j.critrevonc.2010.01.010 (2010).
Rupnarain, C., Dlamini, Z., Naicker, S. & Bhoola, K. Colon cancer: genomics and apoptotic events. (2004).
Rieger, M. A., Parlesak, A., Pool-Zobel, B. L., Rechkemmer, G. & Bode, C. A diet high in fat and meat but low in dietary fibre increases the genotoxic potential offaecal water’. Carcinogenesis 20, 2311–2316 (1999).
Benson, A. B. et al. NCCN guidelines insights: Colon cancer, version 2.2018. J. Natl. Compr. Canc. Netw. 16, 359–369. https://doi.org/10.6004/jnccn.2018.0021 (2018).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. https://doi.org/10.1016/j.cell.2012.03.042 (2012).
Niu, X., Chen, L., Li, Y., Hu, Z. & He, F. Seminars in Cancer Biology (Elsevier, 2022).
Friedmann Angeli, J. P., Krysko, D. V. & Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19, 405–414. https://doi.org/10.1038/s41568-019-0149-1 (2019).
Liang, H. et al. Ferroptosis open a new door for colorectal cancer treatment. Front. Oncol. 13, 1059520. https://doi.org/10.3389/fonc.2023.1059520 (2023).
Zhang, Y. & Xie, J. Targeting ferroptosis regulators by natural products in colorectal cancer. Front. Pharmacol. 15, 1374722. https://doi.org/10.3389/fphar.2024.1374722 (2024).
Rus Bakarurraini, N. A. A., Ab Mutalib, N. S., Jamal, R. & Abu, N. The landscape of tumor-specific antigens in colorectal cancer. Vaccines 8, 371 (2020).
Yang, C., Zhang, Y., Xu, X. & Li, W. Molecular subtypes based on DNA methylation predict prognosis in colon adenocarcinoma patients. Aging 11, 11880–11892. https://doi.org/10.18632/aging.102492 (2019).
Konstorum, A. et al. Systems biology of ferroptosis: A modeling approach. J. Theor. Biol. 493, 110222. https://doi.org/10.1016/j.jtbi.2020.110222 (2020).
Gui, C. P. et al. Identification of mRNA vaccines and conserved ferroptosis related immune landscape for individual precision treatment in bladder cancer. J. Big Data 9, 88. https://doi.org/10.1186/s40537-022-00641-z (2022).
Wu, B., Zhang, B., Li, B., Wu, H. & Jiang, M. Cold and hot tumors: from molecular mechanisms to targeted therapy. Sign. Transduct. Target. Ther. 9, 274. https://doi.org/10.1038/s41392-024-01979-x (2024).
Xia, W. et al. The role of ferroptosis in colorectal cancer and its potential synergy with immunotherapy. Front. Immunol. 15, 1526749. https://doi.org/10.3389/fimmu.2024.1526749 (2024).
Cui, K., Wang, K. & Huang, Z. Ferroptosis and the tumor microenvironment. J. Exp. Clin. Cancer Res. 43, 315. https://doi.org/10.1186/s13046-024-03235-0 (2024).
Xie, Y. et al. Ferroptosis: process and function. Cell Death Differ. 23, 369–379. https://doi.org/10.1038/cdd.2015.158 (2016).
Zhang, Q. et al. The classification and prediction of ferroptosis-related genes in ALS: a pilot study. Front. Genet. 13, 919188. https://doi.org/10.3389/fgene.2022.919188 (2022).
Dai, X., Zhang, R. & Wang, B. Contribution of classification based on ferroptosis-related genes to the heterogeneity of MAFLD. BMC Gastroenterol. 22, 55. https://doi.org/10.1186/s12876-022-02137-9 (2022).
Poursaitidis, I. et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 18, 2547–2556. https://doi.org/10.1016/j.celrep.2017.02.054 (2017).
Xu, Y. et al. Inhibition of the RBMS1/PRNP axis improves ferroptosis resistance-mediated oxaliplatin chemoresistance in colorectal cancer. Mol. Carcinog. 63, 224–237. https://doi.org/10.1002/mc.23647 (2024).
Bernig, T., Richter, N., Volkmer, I. & Staege, M. S. Functional analysis and molecular characterization of spontaneously outgrown human lymphoblastoid cell lines. Mol. Biol. Rep. 41, 6995–7007. https://doi.org/10.1007/s11033-014-3587-6 (2014).
Yu, J. et al. RBMS1 suppresses colon cancer metastasis through targeted stabilization of its mRNA regulon. Cancer Discov. 10, 1410–1423. https://doi.org/10.1158/2159-8290.CD-19-1375 (2020).
Dankert, J. T., Wiesehofer, M., Wach, S., Czyrnik, E. D. & Wennemuth, G. Loss of RBMS1 as a regulatory target of miR-106b influences cell growth, gap closing and colony forming in prostate carcinoma. Sci. Rep. 10, 18022. https://doi.org/10.1038/s41598-020-75083-9 (2020).
Lou, S. et al. Comprehensive characterization of RNA processing factors in gastric cancer identifies a prognostic signature for predicting clinical outcomes and therapeutic responses. Front. Immunol. 12, 719628. https://doi.org/10.3389/fimmu.2021.719628 (2021).
Zhang, J. et al. Loss of RBMS1 promotes anti-tumor immunity through enabling PD-L1 checkpoint blockade in triple-negative breast cancer. Cell Death Differ. 29, 2247–2261. https://doi.org/10.1038/s41418-022-01012-0 (2022).
Lin, X. L. et al. Overexpression of NOX4 predicts poor prognosis and promotes tumor progression in human colorectal cancer. Oncotarget 8, 33586–33600. https://doi.org/10.18632/oncotarget.16829 (2017).
Zhu, J. et al. NOX4 suppresses ferroptosis through regulation of the pentose phosphate pathway in colorectal cancer. Curr. Med. Sci. 45, 264–279. https://doi.org/10.1007/s11596-025-00013-7 (2025).
Shen, C. J. et al. Oleic acid-induced NOX4 is dependent on ANGPTL4 expression to promote human colorectal cancer metastasis. Theranostics 10, 7083–7099. https://doi.org/10.7150/thno.44744 (2020).
Cho, S. Y. et al. Expression of NOX family genes and their clinical significance in colorectal cancer. Dig. Dis. Sci. 63, 2332–2340. https://doi.org/10.1007/s10620-018-5121-5 (2018).
Zhang, Y. et al. High expression of FABP4 in colorectal cancer and its clinical significance. J. Zhejiang Univ. Sci. B 22, 136–145. https://doi.org/10.1631/jzus.B2000366 (2021).
Woischke, C. et al. CYB5R1 links epithelial-mesenchymal transition and poor prognosis in colorectal cancer. Oncotarget 7, 31350–31360. https://doi.org/10.18632/oncotarget.8912 (2016).
Zheng, J. & Conrad, M. Ferroptosis: when metabolism meets cell death. Physiol. Rev. 105, 651–706. https://doi.org/10.1152/physrev.00031.2024 (2025).
Zhang, S. et al. CPEB1 controls NRF2 proteostasis and ferroptosis susceptibility in pancreatic cancer. Int. J. Biol. Sci. 20, 3156–3172. https://doi.org/10.7150/ijbs.95962 (2024).
Mauri, G., Arena, S., Siena, S., Bardelli, A. & Sartore-Bianchi, A. The DNA damage response pathway as a land of therapeutic opportunities for colorectal cancer. Ann. Oncol. 31, 1135–1147. https://doi.org/10.1016/j.annonc.2020.05.027 (2020).
Zhu, R. et al. Ferroptosis-related genes with post-transcriptional regulation mechanisms in hepatocellular carcinoma determined by bioinformatics and experimental validation. Ann. Transl. Med. 10, 1390 (2022).
Luo, L., Yao, X., Xiang, J., Huang, F. & Luo, H. Identification of ferroptosis-related genes for overall survival prediction in hepatocellular carcinoma. Sci. Rep. 12, 10007 (2022).
Yang, C., Huang, S., Cao, F. & Zheng, Y. Role of ferroptosis-related genes in prognostic prediction and tumor immune microenvironment in colorectal carcinoma. PeerJ 9, e11745. https://doi.org/10.7717/peerj.11745 (2021).
Yue, Q. et al. Classification of colorectal carcinoma subtypes based on ferroptosis-associated molecular markers. World J. Surg. Oncol. 20, 117. https://doi.org/10.1186/s12957-022-02575-5 (2022).
Funding
This work was supported by the National Natural Science Foundation Cultivation Project [No.210208], the Scientific Research Translational Foundation of Wenzhou Safety (Emergency) Institute of Tianjin University [No. TJUWYY2022025], and Chinese Anti-Cancer Association—HengRui Anti-angiogenesis Targeted Tumor Research Fund [No.2021001045].
Author information
Authors and Affiliations
Contributions
Dalu Kong conceived and designed the study, Jun Hu, Jian Li and Yixuan Dong collected data and wrote the manuscript. Wenpeng Wang and Xin Yue prepared the figures. Hongbo Zhang analyses the data. All authors have read the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, J., Li, J., Dong, Y. et al. Identification of ferroptosis related genes and subtypes in colorectal cancer. Sci Rep 15, 22717 (2025). https://doi.org/10.1038/s41598-025-08901-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-08901-7
