
Abstract
Low-penetrance pathogenic copy number variations (CNVs), variation of uncertain significance (VUS) CNVs and likely pathogenic CNVs present a challenge for prenatal diagnosis. Previous studies have clarified the influence of a parent-of-origin test on the prenatal VUS CNVs. However, the influence of parent-of-origin tests on prenatal likely pathogenic (LP) or low-penetrance pathogenic CNVs (pCNVs) have not been evaluated. Here, among 2273 pregnant women undergoing prenatal diagnosis, 236 CNVs were reported by chromosomal microarray analysis (CMA) including 69 full-penetrance pCNVs, 44 low-penetrance pCNVs, 113 VUS CNVs and 10 LP CNVs. Based on the subsequent parent-of-origin tests, CNVs were classified as de novo, inherited and unknown group. Firstly, a total of 112 couple (62 VUS CNVs, two LP CNVs and 48 pCNVs) chose parent-of-origin tests and 88 inherited CNVs (51 VUS CNVs, two LP CNVs and 35 pCNVs ) were identified. Then, the effect of parent-of-origin tests was focused on 44 low-penetrance pCNVs, 113 VUS CNVs and 10 LP CNVs in this study (n = 167). For 44 low-penetrance pCNVs, termination of pregnancy (TOP) rates in de novo, inherited and unknown group were 100% (5/5), 23.5% (4/17) and 40.9% (9/22), respectively. TOP decisions in low-penetrance pCNVs were mainly affected by de novo and abnormal ultrasound findings. For 113 VUS CNVs, inherited VUS CNVs dramatically reduced anxiety reflected by TOP rates in de novo (18.2%, 2/11), inherited (0/51) and unknown group (2.0%, 1/51). Notably, prenatal minor structural defects often disappeared after birth. These results suggested the majority VUS CNVs have no appreciable pathogenicity. For 10 LP CNVs, TOP rates in inherited and unknown group were 0% (0/2) and 87.5% (7/8), which suggested that it is imperative that parent-of-origin tests be offered for LP CNVs to bring the classification to pathogenic or VUS.
Similar content being viewed by others

Introduction
Copy number variations (CNVs) refer to a deletion or duplication of DNA segments that are typically larger than 1 KB in size. Genomic regions involved in CNVs may be “junk” and these CNVs were observed with a high frequency in the general population, suggesting that these CNVs are not rare but polymorphic variations. On the contrary, genomic content contained by CNVs may be important and dosage sensitive, and this indicates that these CNVs are deleterious1. Great efforts have been made in understanding and analyzing the effects of CNVs. American College of Medical Genetics and Genomics (ACMG) has developed consistent methods of evaluating the genomic content of a CNV region and correlating clinical findings with those reported in the medical literature, with the ultimate goal of producing consistent, evidence-based clinical classification across laboratories in 2015, and these methods were updated in 2020 1. According to ACMG guidelines, CNVs with a final point value ≥ 0.99 or between 0.90 and 0.98 are considered pathogenic and likely pathogenic (LP) separately. The variant of uncertain significance (VUS) corresponds to points between − 0.89 and 0.89, while refuting evidence arriving at scores between − 0.90 and − 0.98, or ≤ − 0.99 are considered likely benign (LB) and benign, respectively.
With the rapid advancement of genetic technology, many pathogenic CNVs (pCNVs) such as distal 1q21.1 deletion/duplication (includes GJA5), 22q11.2 deletion syndrome (DGS/VCFS), and 22q11.21 duplication syndrome (cat eye syndrome)3,4,5 have been found. Nevertheless, in the clinical setting, some pCNVs were often inherited from unaffected carriers, and the CNVs were also observed in the general population at a relatively high frequency. This phenomenon can be partially explained by incomplete penetrance and variable expressivity. For example, the expression of any phenotype associated with 15q11.2 microdeletion has been estimated to be 10.4%6,7. As well as 15q11.2 microdeletion, the established low-penetrance was also observed in distal 1q21.1 deletion/duplication (includes GJA5) (36.9%/29.1%), 16p11.2 deletion/duplication (includes TBX6) (46.8%/27.2%), 17q12 deletion/duplication (HNF1B) (34.3%/21.1%), and 22q11.21 duplication (includes TBX1) (21.9%)7. Incomplete penetrance may be caused by a range of factors including variants in regulatory regions, epigenetics, and environmental factors8.
The dosage sensitivity score of definite pCNVs is three. However, definite dosage sensitivity regions and genes are very limited. For example, only about 450 definite haploinsufficiency (HI)/ Triplosensitivity (TS) genes or regions are documented by ClinGen. Most CNVs were classified as VUS that represents a broad category1. Examples of VUS may include: (1) the size of CNVs exceeds the fragment threshold defined by the laboratory for reporting but does not contain encoding genes; (2) conflicting associations between CNVs with clinical phenotype described in published literature and genomics databases; (3) microdeletion contains a small number of genes or within an individual gene causing autosomal dominant (AD) diseases with unestablished pathogenic mechanism such as gain of function (GOF), loss of function (LOF), dominant negative effect9,10. At present, chromosomal microarray analysis (CMA) and copy number variation sequencing (CNV-seq) are widely used in prenatal diagnosis, and both have returned a 1-7.7% higher yield of clinically relevant CNVs11,12. As expected, CMA and CNV-seq also identified additional about 1.7–3.4% VUS CNVs12,13.
In addition, LP CNVs are often reported by using the evidence scoring metric. In general, these variants have strong evidence to suggest that they will ultimately be determined to be disease-causing, but not enough yet to definitively assert pathogenicity. Examples of LP CNVs may include: (1) deletions involving the 5’ end (plus additional coding sequence) of established HI genes; (2) deletions involving multiple exons (through the 3’ end of the gene) in an established HI gene ; (3) deletions or duplications involving genes with multiple case reports reported in consistent, highly specific phenotype; and (4) number of protein-coding genes wholly or partially included in the copy number loss > = 35 + genes or in the copy number gain > = 50 + genes1.
In prenatal period, the findings of low-penetrance pathogenic, VUS and LP CNVs present a challenge for counseling and cause anxiety. In our study, among 2273 pregnant women undergoing prenatal diagnosis, 44 low-penetrance pCNVs, 113 VUS CNVs and 10 LP CNVs were detected by chromosomal microarray analysis (CMA). Therefore, we performed a retrospective study to explore the significance of a parent-of-origin test on 44 low-penetrance pCNVs, 113 VUS CNVs and 10 LP CNVs, thereby improving the prenatal counseling and manage for these three types CNVs.
Results
Fetal CNVs clinical significance distribution and origin
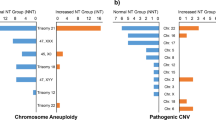
In total, 2273 amniotic fluid samples were retrospectively analyzed, and 236 CNVs of these samples were detected, including 113 VUS CNVs (5.0%, 113/2273), 10 LP CNVs (0.4%, 10/2273) and 113 pCNVs (5.0%, 113/2273). There were 44 low-penetrance pCNVs among 113 pCNVs (Fig. 1).

The distribution of 236 CNVs.
124 couples refused a parent-of-origin test while 112 couple agreed. Among VUS CNVs, de novo and inheritance account for 17.5% (11/62) and 82.3% (51/62), respectively (Fig. 1). Among pCNVs, de novo and inheritance represent 27.1% (13/48) and 72.9% (35/48), respectively (Fig. 1). Among LP CNVs, de novo and inheritance make up 0% (0/2) and 100% (2/2),separately (Fig. 1). After a parent-of-origin test, the clinical interpretation of 51 inherited VUS CNVs changed and tended to be LB and the rate of VUS CNVs decreased to 2.7% (62/2273) .
Among 88 inherited CNVs, maternal origin CNVs constitute a large fraction (73.9%, 65/88) while paternal origin CNVs represent a small percentage (26.1%, 23/88). To avoid maternal CNVs detected by NIPT introducing a significant bias, the 30 prenatal VUS CNVs and 28 pCNVs with prenatal diagnosis indications as a high-risk result from NIPT were excluded. Among the remaining 39 inherited CNVs, maternal origin accounts for 66.7%, whereas paternal origin consists of 33.3%. For inherited VUS CNVs, maternal origin (48%) and paternal origin (52%) have a proximity in proportion. However, for inherited LP CNVs and pCNVs, maternal origin both make up a 100% proportion (Fig. 1).
Ultrasound findings and pregnant outcomes in de novo, inherited and unknown low-penetrance pCNVs
For low-penetrance pCNVs, the case number in de novo, inherited and unknown group is 5, 17 and 22, respectively (Fig. 2). Primary clinical indications, CNVs, origin and pregnant outcomes are listed in Supplementary Table 1.
The rate of abnormal ultrasound findings in de novo, inherited and unknown group was 100% (5/5), 11.8%(2/17) and 36.36%(8/22), respectively (Fig. 2).
Five de novo cases were all found with minor structural abnormalities by ultrasound, including dysplasia of nasal bone, ventricular bright spots and single umbilical artery, increased renal echogenicity, and thickened nuchal translucency (NT). All pregnancies were terminated (100%, 5/5) (Table 1) (Fig. 2).

The influence of parent-of-origin tests on pregnant decisions and outcomes in fetuses with low-penetrance pCNVs. Based on parent-of-origin tests, the cases number in de novo, inherited and unknown group was 5, 17 and 22, respectively. The total TOP rate among low-penetrance pCNVs was 40.9% (18/44). The TOP rates of de novo, inherited and unknown group were 100%, 23.5% and 40.9%, respectively. Among 26 cases with continued pregnancy, two times follow-up were performed.
Of 17 inherited cases, there were two cases presenting structure malformations including omphalocele and diaphragmatic hernia. After genetic counseling, TOP decisions were made by the couple (Table 1). Another two carrier mothers whose pregnancies were continued showed disease phenotype including epilepsy and intellectual disability. After birth, normal development was reported by their parents. Among the remaining 13 inherited cases with normal prenatal ultrasound, two pregnancies were terminated and reasons were listed in Table 1. The total termination of pregnancy (TOP) rate was 23.5% (4/17) (Fig. 2).
For 22 unknown origin cases, there were eight cases presenting with abnormal ultrasound containing right aortic arch, widened ventricles, nasal bone dysplasia, restricted growth, thickened nuchal fold, brain dysplasia, enhanced renal echogenicity and permanent right umbilical vein, five of which were terminated (Table 1). It is noteworthy that one case was found with enlargement of the brain ventricles at 23 weeks firstly. However, re-examination showed hypoplasia of corpus callosum at 28 weeks. The pregnancy was terminated and WES was performed. A pathogenic missense variant c.719 C > T (p.Pro240Leu) in L1CAM was screened out via WES. Among the remaining 14 cases with normal prenatal ultrasound, four were terminated and reasons were listed in Table 1. The rate of TOP was 40.9% (9/22) (Fig. 2).
All 18 cases with TOP decisions were listed in Table 1. The TOP rate of low-penetrance pCNVs was 40.9% (18/44).
A total of 26 pregnant women continued with their pregnancies. The first follow-up was routinely performed when infants were ≥ 6 and < 12 months old. The percentages of abnormal pregnancy outcomes (pregnancy loss, preterm delivery, neonatal death, birth defects) and delayed development at six months old in the inherited and unknown group were all 0%.
Since the phenotype caused by low-penetrance pCNVs are often nonspecific and neurological. These nonspecific phenotype are hard to be noticed during early childhood periods. Thus, cases with low-penetrance pCNVs were followed up again before publication. In the inherited group, eight cases being 24 ≤ age < 48 months old and five cases being 12 ≤ age < 24 months old all showed normal development. In unknown group, six cases were 12 ≤ age < 24 months old and one case showed delayed language and motor development when he was nearly 14 months old. Six cases being 24 ≤ age < 48 months old all showed normal development (Fig. 2).
Ultrasound findings and pregnant outcomes in de novo, inherited and unknown VUS CNVs
The number of VUS cases in de novo, inherited and unknown group was 11, 51 and 51, respectively (Fig. 3). Primary clinical indications, CNVs, disease-related genes, ACMG score and classification, origin, pregnant outcomes and follow-up were listed in Supplementary Table 1.

The influence of parent-of-origin tests on pregnant decisions and outcomes in fetuses with VUS CNVs. Based on parent-of-origin tests, the cases number in de novo, inherited and unknown group was 11, 51 and 51, respectively. The total TOP rate of VUS CNVs cases was 2.7% (3/113). The TOP rates of de novo, inherited and unknown group were 18.2%, 0% and 2%, respectively. Among 110 cases with continued pregnancy, routine follow-up was performed. The abnormal pregnancy outcomes in the de novo, inherited and unknown group were listed.
The percentage of abnormal ultrasound findings in de novo, inherited and unknown group was 36.4%(4/11), 31.4%(16/51) and 51%(26/51), separately.
Ultrasound findings and pregnancy outcomes in de novo VUS CNVs
Of 11 de novo VUS CNVs, there were four cases showing abnormal ultrasound including three cases with thickened NT and one case carrying a 672.4Kb microdeletion in 10q26.3(134,753,928 − 135,426,386) region with minor structure abnormalities (mild pericardial effusion). This CNV was observed in normal population database. Furthermore, genes involved in were not apparently related to the effusion after detailed analysis (Supplementary Table 1). After genetic counseling, the couple determined to continue the pregnancy since mild pericardial effusion could be treated surgically after birth. Through follow-up, this fetus was born at 37 weeks and showed normal development. Among the remaining seven cases with normal ultrasound, two cases were abandoned voluntarily even without abnormal ultrasound findings (18.2%, 2/11) (Fig. 2). For nine continued pregnancies, no birth defects and delayed development were reported by their parents.
Ultrasound findings and pregnancy outcomes in inherited VUS CNVs
Of 51 inherited VUS CNVs, 16 cases consisting of 10 cases with abnormal soft marker and six cases showing minor ultrasound structural abnormalities were found with prenatal abnormal ultrasound. Of the 10 cases with abnormal soft marker, one case with a 660Kb microdeletion in 22q11.21(20,716,876 − 21,381,242) region presented renal and ureter abnormalities after birth. Renal and ureter abnormalities have been reported in many individuals with 22q11.21(20,716,876 − 21,381,242) deletion. A parent-of-origin test confirmed that this microdeletion was inherited from asymptomatic mother, supporting the incomplete penetrance. The other nine all showed full-term birth without any anomalies. For the six cases showing minor ultrasound structural defects including cervical cystic hygroma, mild tricuspid regurgitation, mild ventricular septal defect, and single umbilical artery, WES testing were also recommended to exclude other genetic causes but were refused. These six pregnancies were continued since the above mentioned malformations rarely lead to function defects. By being followed up, all six cases showed full-term birth and all minor ultrasound structural defects disappeared. These results confirmed that VUS CNVs were not the genetic cause of minor ultrasound structural defects. Of the remaining 35 inherited CNVs without abnormal imaging findings, one case was born with clubfoot and hypotonia and died shortly after birth. Trio-WES was performed and the two pathogenic NEB variant in trans were detected.
Inherited VUS CNVs involving multiple exons of HI/TS genes either with established or emerging evidence, and unreported in population databases
Of 51 inherited VUS CNVs, four inherited VUS CNVs were not reported in population databases but involves multiple exons of HI/TS genes either with established or emerging evidence such as TP63, NSDHL and DMD. In three cases carrying maternal original CNVs involving partial exons of established HI genes DMD and NSDHL in X chromosome, a familial segregation analysis was performed in fathers or brothers of the pregnant women. Eventually, CNVs were detected in unaffected male carriers, ruling out the pathogenicity. In another maternal microdeletion inside in exon 2–4 of HI gene TP63 with emerging evidence, a gene associated with dermatosis, dry and flaky skin were observed in both carrier mother and the proband. However, unaffected carrier was also found in this family, supporting potential incomplete penetrance of TP63.
Association analysis between inherited VUS CNVs and carrier parents phenotype
In inherited group, there was one carrier mother affected by cleft lip (Supplementary Table 1). After the analysis of the database, associations between the identified CNV or involved genes and cleft lip were unreported so far. Follow-up information verified that fetus was born with full term without visible defects.
Ultrasound findings and pregnant outcomes in unknown VUS CNVs
Of 51 unknown VUS CNVs, there were 26 cases, one of which was terminated, with abnormal imaging findings including 17 with abnormal soft marker and nine cases with minor structure defects. The follow-up results showed that there were three cases born with minor lesion of congenital heart disease and that one case with poor intestinal rotation. The remaining 25 pregnancies without abnormal image findings were continued and the fetuses were born normally.
The total TOP rate of VUS CNVs was 2.7% (3/113) (Fig. 3). A total of 110 pregnant women continued with their pregnancies (Fig. 3). All cases showed normal motor and language development at ≥ 6 and < 12 months old except for one died due to NEB variant (Fig. 3).
Ultrasound findings and pregnant outcomes in de novo, inherited and unknown LP CNVs
The number of cases with LP CNVs in de novo, inherited and unknown group was 2, 0 and 8, respectively (Fig. 4). Primary clinical indications, CNVs, ACMG scores, origin and pregnant outcomes were listed in Supplementary Table 1.

The influence of parent-of-origin tests on pregnant decisions and outcomes in fetuses with LP CNVs. Based on parent-of-origin tests, the cases number in de novo, inherited and unknown group was 0, 2 and 8, respectively. The total TOP rate of LP CNVs cases was 70% (7/10). The TOP rates in inherited and unknown group were 0% and 87.5%, respectively. Among eight cases with continued pregnancy, routine follow-up was performed. The abnormal pregnancy outcomes in inherited and unknown group were listed.
For two inherited LP CNVs, both couples referred to prenatal diagnosis due to high-risk results from NIPT. Subsequently, CMA revealed a 9.4 Mb deletion in 10q11.22q21.1(46,141,836 − 55,602,540) region and 33.2 Mb duplication in 13q21.1q31.3(58,366,953 − 91,571,505) region, respectively. For the case with deletion in 10q11.22q21.1, the CNV was inherited from unaffected mother. Similar variation, inherited from unaffected parents, had been reported in individuals who presented non-specific phenotype like intellectual disability and delayed development. All these evidence indicated the incomplete penetrance. For another case with duplication in 13q21.1q31.3, the CNV was inherited from mother affected by cleft lip. Both two families denied disease family history and rejected to expand family pedigree verification. After genetic counseling, both couples decided to continue their pregnancies. The follow-ups showed that both were born in full-term, and no delayed development and visible defect were noticed.
For eight unknown LP CNVs, seven were terminated and TOP rate was 87.5%. For the one continued pregnancy, 18.8 Mb duplication in 4q34.1q35.2(172,142,544 − 190,957,460) region and 5.3 Mb deletion in 9p24.3p24.1(208,454-5,508,647) region were detected. The first follow-up after birth showed that the case suffered from minor lesion of congenital heart disease. The second follow-up at 10 months old showed delayed development. For example, he could not sit well independently.
Discussion
In the present study, 236 CNVs consisting of 113 VUS CNVs, 10 LP CNVs and 113 pCNVs were detected. In a previous study, a trio analysis of CNVs in 141 cases showed that 72.3% of fetal CNVs were derived from parents, and 27.7% of these cases were de novo variations14. Similarly, in our study including 112 parent-of-origin tests, the overwhelmingly high proportion of inherited CNVs among which VUS CNVs was 82.3% (51/62), LP CNVs was 100% (2/2) and pCNVs was 72.9%(35/48) were also observed (78.6%, 88/112).
Previous studies have reported the influence of the parent-of-origin tests on clinical interpretation of fetuses with VUS CNVs. VUS CNVs in fetuses may be downgraded to LB if trio analysis showed that the VUS CNVs were inherited from healthy parents. In their study, Lin et al. reported a 5.2% VUS, which decreased to 1.9% (265/14073) after parent-of-origin tests15. In another study with 102 parent-origin test cases, the number of VUS was changed from 62 to 1214. The proportions of VUS CNVs before and after parent-of-origin tests in our study was 5.0% and 2.7%, respectively. Potential reasons of higher VUS rate in our study included: (1) invasive tests were performed in some VUS CNVs indicated by NIPT; (2) only 54.9% (62/113) VUS cases determined a parent-of-origin test, mainly owing to high costs and long time consuming; (3) the lack of CNV database based on Chinese population leads to failing recognition of common polymorphisms CNVs. In the future, the increased speed along with decreasing cost of sequencing will result in a higher acceptance rate of parent-of-origin test. A big multi-center CNV database on the basis of Chinese population should be encouraged, which will help to identify common polymorphisms.
A previous study including 141 families reported that 59.8% of fetal CNVs were maternal origin and 40.2% were paternal origin14. This study is consistent with the result in another two studies that a higher ratio of maternal sources than paternal origin was reported15,16. Researchers pointed that fetuses acquired more maternal CNVs than paternal CNVs and it is necessary for clinicians to collect detailed information about maternal phenotypes16. In our study, uneven transmission ratio of inherited CNVs, 73.9% (65/88) maternal origin and 26.1% (23/88) paternal origin, was consistent with above researches. To avoid maternal CNVs detected by NIPT introducing a significant bias, the 30 VUS CNVs cases and 28 pCNVs cases with prenatal diagnosis indications as high-risk results from NIPT were excluded. Surprisingly, uneven transmission in VUS CNVs disappeared (48% vs. 52%), but remained in pCNVs (maternal origin 100% VS paternal origin 0%) (Fig. 1).
The dosage sensitivity score of low-penetrance pCNVs is three, which indicates that these CNVs are pathogenic. Some clinicians would claim that the pathogenic conclusion should be highlighted on the report and the parent-of-origin test will not change the grade and clinical interpretation of low-penetrance pCNVs, while others hold that low-penetrance pCNVs should be interpreted as VUS. In this study, TOP rates for pCNVs with low-penetrance was 40.9% (18/44), which is greatly higher than VUS CNVs (2.7%, 3/113). Our data suggested that difference of pregnant decisions between low-penetrance pCNVs and VUS CNVs was great. A known pathogenic CNV characterized by incomplete penetrance cannot be classified as VUS. Instead, its clinical significance might be uncertain due to lower penetrance or variable expression, highlighting the importance of family studies for interpretation.
Currently, our research was the first study analyzing the influence of the parent-of-origin test on pregnancy outcomes of fetal low-penetrance pCNVs. Here, the proportion of abnormal ultrasound findings in de novo, inherited and unknown group was 100% (5/5), 11.8%(2/17) and 36.36%(8/22), respectively. The TOP rates in de novo, inherited and unknown group were 100% (5/5), 23.5%(4/17) and 40.9% (9/22), respectively. This result indicates that fetus carrying de novo low-penetrance pCNVs is more likely to be affected. Among 18 cases with TOP, 12 cases were with abnormal ultrasound screening. Therefore, TOP decisions in low-penetrance pCNVs were mainly affected by de novo and abnormal ultrasound findings.
For the TOP rate in VUS CNVs, a previous study showed significant difference in the de novo, inherited and unknown groups (47.7%,1.7%,14.3%)15. In our study, those figures were 18.2%, 0% and 2.0%, respectively. These results suggested that a parent-of-origin test has great effects on the TOP rate of VUS CNVs, and inherited VUS CNSs dramatically reduce anxiety. Although the TOP rate of inherited VUS CNVs was extremaly low both in our and other studies, there are some points should be emphasized: (1)re-examination of ultrasound regularly to help evaluate the fetus status; (2) reviewing the database to explore the relationship between CNVs and phenotype; (3) when VUS CNVs show potential potential incomplete penetrance or may disrupt vital genes such as DMD and TP63 in our study, family segregation analysis should be further expanded; (4)for severe structural defects, WES should be recommended.
In our study, the 110 pregnancies with VUS CNVs were continued. The parent-of-origin test results, health information of carrier parents and follow-up results were recorded in detail, thus providing rich and valuable data and facilitates genetic counseling. For instance, expanding pedigree verification of VUS CNVs involving NSDHL and DMD rules out its pathogenicity, whilst VUS CNVs involving TP63 indicates its potential incomplete penetrance. In addition, some VUS CNVs were frequently observed in our data such as a deletion in 2q13(110498142–110980295) involving NPHP1, a duplication in 2p12(78631710_79851089) involving CTNNA2, a duplication in Xp22.31(6,449,836-8,135,644) involving STS, a duplication in 13q12.12(23,554,650 − 24,826,638) involving SGCG and SACS, and a deletion in 5q12.1(59900364–60277057) containing ERCC8 and NDUFAF2.
It has been estimated that the incidence of birth defects is ~ 5.6% in China and ~ 6% worldwide17. Chen et al. showed that among 721 fetuses with VUS CNVs, the rate of abnormal pregnancy outcomes in de novo, inherited and unknown origin groups was 6.2%, 4.3% and 6.6%, respectively, and the total rate of abnormal pregnancy outcomes was 5.1%15. Shi et al. reported that among 139 pregnancies with VUS CNVs, only 5% of the liveborn by 2–4 years of age had signs of diseases, none of which have been previously reported to be associated with the identified VUS13. In our study including 113 VUS CNVs, birth defects were often mild and treatable. It is noticeable that prenatal minor ultrasound structural defects often disappeared after birth. Above studies suggested that the proportion of birth defects in fetuses with VUS CNVs is close with the overall incidence of birth defects in China. The majority of VUS CNVs has no appreciable pathogenicity.
For the 10 LP CNVs, only two parent-of-origin tests were performed. Among the eight unknown cases, seven were terminated and continued one was born with birth defects, and this reflected that the importance of parent-of-origin test was not fully clarified. For LP CNVs, it needs to be emphasized that parent-of-origin test should be performed to bring the classification to pathogenic or VUS.
In conclusion, it is imperative that parent-of-origin tests be offered for families with low-penetrance pathogenic, LP and VUS CNVs. Apart from altering pregnant decisions, parent-of-origin results will provide more solid evidence for evaluating the potential effect of these CNVs. However, limitations in this study need to be acknowledged. The number of low-penetrance pCNVs identified in our study was small and 52.5% (124/236) couples refused a parent-of-origin test, making our conclusions biased.
Materials and methods
The study was approved by Ethics Committee of Jiangsu Huai’an Maternity and Child Health Care Hospital (2021042). All methods were performed in accordance with the relevant guidelines and regulations.
Local guidelines regarding invasive prenatal testing
Primary clinical indications for a invasive prenatal testing in our hospital was recommended based on a joint consensus “Application of copy number variation sequencing for prenatal diagnosis” and were listed as follows:
-
(1)
Abnormal ultrasound findings including soft markers and structure.
-
(2)
A high-risk result indicated by noninvasive prenatal testing (NIPT) including autosomal aneuploidies, sex chromosomes aneuploidies, CNVs fully containing regions or genes with established haploinsufficiency or triplosensitivity (HI/TS = 3).
-
(3)
A high-risk result from maternal serum screening.
-
(4)
A poor gestation and birth history.
-
(5)
Advanced maternal age (age ≥ 35).
-
(6)
A parent with chromosomal rearrangements.
In addition, based on informed consent, some VUS CNVs indicated by NIPT in special circumstances will also be recommended for further prenatal diagnosis, which were listed as follows:
-
(7)
A VUS CNV indicated by NIPT partially overlaps the gene or region with established haploinsufficiency or triplosensitivity (HI/TS = 3), which may disrupt the expression of protein and needs to be further confirmed by CMA, such as DMD and TSC1/2.
-
(8)
A VUS CNV indicated by NIPT involves genes or regions with little or emerging evidence for HI or TI ( HI/TS score = 1 or 2). For instance, this CNV was detected in some affected cases with overlapping phenotype but the the case number not big enough or incomplete penetrance was occasionally observed.
Subjects
From January 2018 to August 2022, 2273 pregnant women were referred to the Department of Genetics and Prenatal Diagnosis of the Jiangsu Huai’an Maternity and Child Health Care Hospital for amniocentesis and chromosome testing. All subjects signed an informed consent. The age of the pregnant women ranged from 19 to 45 years old (median, 29). The gestational age ranged from 18 to 33+ 1 weeks (median, 20). Primary clinical indications for prenatal diagnosis included ultrasound abnormality (1004, 44.2%), high-risk results of NIPT (595, 26.2%), high-risk results of maternal serum screening at second trimester (90, 4%), an adverse pregnancy history (211, 9.3%), advanced maternal age (age ≥ 35 years) (117, 5.1%), and other (256, 11.3%). Cases with multiple indications was not counted repeatedly.
Amniocentesis and chromosome testing
Amniocentesis
Amniocentesis was performed by needle puncture of the amnion and 30 mL of amniotic fluid was removed by aspiration. Amniotic fluid was centrifugated in 1500 rpm for 10 min. Deposits were washed by PBS and extracted for genomic DNA by Thermo Fisher MagMAX™ DNA Multi-Sample Ultra 2.0 Kit (A36570, Thermo Fisher). 21 short tandem repeat (STR) markers (labeled with FAM: D2S441, D5S818, D19S433, FGA, D6S1043; labeled with NED: TH01, D8S1179, D18S51, Penta D; labeled with HEX: TPOX, D16S539, D12S391, D10S1248, D2S1338, Penta E; labeled with ROX: AMEL, vWA, D7S820, D13S317, D1S1656, CSF1PO) were used as a quality control to detect any maternal DNA contamination in the fetal samples.
Chromosome testing
During the study, all fetal DNA for prenatal diagnosis tests was hybridized with Affymetrix CytoScan 750 K array by following the manufacturer’s protocol. The chip data in CEL file were analyzed by Chromosome Analysis Suite (CHAS) software (v2.0) to produce CYCHP file. The CNVs in CYCHP file were called by CHAS.
CNVs analysis
CNVs subsequently identified from chromosome plots were queried against public databases, such as DGV v2.0 version (http://dgv.tcag.ca/dgv/app/home), Decipher v9.27(https://www.deciphergenomics.org/), OMIM(https://www.omim.org/), ClinGen(https://www.clinicalgenome.org/), UCSC(https://genome.ucsc.edu/), published papers or the laboratory internal database. Any possible pathogenicity of the CNV for clinical interpretation, was determined according to the 2020 ACMG guidelines. CNVs were classified as benign, LB, VUS, LP or pathogenic. For clinical reporting of patient results, only VUS, LP or pathogenic were considered.
In our study, low-penetrance CNVs refer to 1q21.11q21.1 recurrent deletion (includes GJA5) (penetrance = 36.9%) or duplication (penetrance = 29.1%), 15q11.2 recurrent deletion (includes NIPA1) (penetrance = 10.4%), 16p11.2 recurrent deletion (includes TBX6) (penetrance = 46.8%) or duplication (penetrance = 27.2%), 16p13.11 recurrent region (includes MYH11) (penetrance = 13.1%), 17q12 recurrent deletion (includes HNF1B) (penetrance = 34.3%) or duplication (penetrance = 21.1%), and 22q11.21 duplication (includes TBX1) (penetrance = 21.9%)7. For VUS CNVs, fragment deletion smaller than 500 KB or duplication less than 1000 KB was generally not reported if it was observed in the general population but frequency was lower than 1% or no protein-coding genes were involved.
Parent-of-origin test by CNV-seq
Parent-of-origin test via CNV-seq was recommended for VUS, LP or pathogenic CNV. Genomic DNA extracted from peripheral blood was fragmented to an average size of 180 ~ 280 bp and used to create a DNA library following established Illumina paired-end protocols. The Illumina Novaseq 6000 platform (Illumina Inc., San Diego, CA, USA) was used for genomic DNA sequencing by Novogene Bioinformatics Technology Co., Ltd (Beijing, China) to generate 150-bp paired-end reads. After sequencing, base-call file conversion and demultiplexing were performed with the bcl2fastq software(v2.19) (Illumina). The resulting fastq data were analyzed by Fastp(v0.23.2) to remove low quality reads, and were then aligned to the reference human genome (hg19) using the Burrows-Wheeler Aligner (BWA)(v0.7.17), and duplicate reads were marked using GATK tools(v4.2.6.1). CNVs based on in house controls were called with WisecondorX(v1.2.5) and CNVnator(v0.4.1). The results were filtered and annotated by in house software.
If the fetal VUS CNVs were inherited from health parents, the CNVs were reevaluated as LB. If the fetal VUS CNVs were de novo, the pathogenicity were still VUS.
Phenotypic assessment of carrier parents
Family history of disease, health status and academic degree were inquired in detail. Given the difficulties in identifying non-specific phenotype like autism spectrum disorder (ASD) and behavioral problems, parents carrying low-penetrance pCNVs were required to accepting psychopathology screening by Symptom Checklist 90 (SCL-90) questionnaire. If the total score is ≥ 160 or the positive factor is > 43 or the single factor is ≥ 2, the screening is considered positive.
Follow-up
First follow-up was routinely performed when infants with age ≥ 6 and < 12 months old. The contents of the inquiry mainly included: whether TOP, whether pregnancy loss, whether preterm delivery, the birth parameters (weight, length, and the Apgar score), whether birth defects and six months old development evaluated by Children’s Health Clinic (weight, length, head circumference and corresponding developmental milestones). If they were out of touch, information was checked on the Provincial Maternal and Child Health Information System.
Before publication, cases with low-penetrance pCNVs were followed up again. Developmental milestones including language and motor in cases who were 12 ≤ age < 24 months old and 24 ≤ age < 48 months old were inquired.
Data availability
The datasets generated and/or analysed during the current study are not publicly available due to privacy/ethical restrictions but are available from the corresponding author on reasonable request.
References
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 22, 245–257. https://doi.org/10.1038/s41436-019-0686-8 (2020).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Pang, H. et al. Disorders associated with diverse, recurrent deletions and duplications at 1q21.1. Front. Genet. 11, 577. https://doi.org/10.3389/fgene.2020.00577 (2020).
Portnoi, M. F. Microduplication 22q11.2: A new chromosomal syndrome. Eur. J. Med. Genet. 52, 88–93. https://doi.org/10.1016/j.ejmg.2009.02.008 (2009).
Funato, N. Craniofacial phenotype and Genetics of DiGeorge syndrome. J. Dev. Biol. 10 https://doi.org/10.3390/jdb10020018 (2022).
Cox, D. M. & Butler, M. G. The 15q11.2 BP1-BP2 microdeletion syndrome: A review. Int. J. Mol. Sci. 16, 4068–4082. https://doi.org/10.3390/ijms16024068 (2015).
Rosenfeld, J. A., Coe, B. P., Eichler, E. E., Cuckle, H. & Shaffer, L. G. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet. Med. 15, 478–481. https://doi.org/10.1038/gim.2012.164 (2013).
Kingdom, R. & Wright, C. F. Incomplete penetrance and variable expressivity: From clinical studies to population cohorts. Front. Genet. 13, 920390. https://doi.org/10.3389/fgene.2022.920390 (2022).
Gerasimavicius, L., Livesey, B. J. & Marsh, J. A. Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure. Nat. Commun. 13, 3895. https://doi.org/10.1038/s41467-022-31686-6 (2022).
Backwell, L. & Marsh, J. A. Diverse molecular mechanisms underlying pathogenic protein mutations: Beyond the loss-of-function paradigm. Annu. Rev. Genomics Hum. Genet. 23, 475–498. https://doi.org/10.1146/annurev-genom-111221-103208 (2022).
Wapner, R. J. et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl. J. Med. 367, 2175–2184. https://doi.org/10.1056/NEJMoa1203382 (2012).
Wang, J. et al. Prospective chromosome analysis of 3429 amniocentesis samples in China using copy number variation sequencing. Am. J. Obstet. Gynecol. 219, 287e281–287e218. https://doi.org/10.1016/j.ajog.2018.05.030 (2018).
Shi, P. et al. The uncertainty of copy number variants: Pregnancy decisions and clinical follow up. Am. J. Obstet. Gynecol. https://doi.org/10.1016/j.ajog.2023.01.022 (2023).
Shi, P., Li, R., Wang, C. & Kong, X. Influence of validating the parental origin on the clinical interpretation of fetal copy number variations in 141 core family cases. Mol. Genet. Genomic Med. 7, e00944. https://doi.org/10.1002/mgg3.944 (2019).
Chen, L. et al. Influence of the detection of parent-of-origin on the pregnancy outcomes of fetuses with copy number variation of unknown significance. Sci. Rep. 10, 8864. https://doi.org/10.1038/s41598-020-65904-2 (2020).
Wen, Q., Wang, X., Zhang, H., Liu, X. & Xu, Z. Distribution and transmission of copy number variations of uncertain significance in 105 trios. Mol. Genet. Genomic Med. 10, e (2030). https://doi.org/10.1002/mgg3.2030 (2022).
Zarocostas, J. Serious birth defects kill at least three million children a year. BMJ 332, 256. https://doi.org/10.1136/bmj.332.7536.256-b (2006).
Acknowledgements
We want to express our appreciation to the pregnant women who participated in this study.
Funding
This work was granted by the Maternal and Child Health project of Jiangsu Province (No. F201714 and F201707), Jiangsu Key Laboratory of New Drug Research and Clinical Pharmacy (XZSYSKF2020024), and Huai’an Natural Science Foundation (Grant No. HAB202217).
Author information
Authors and Affiliations
Contributions
Yuefang Liu and Qiong Pan conceived and designed this study. Yuefang Liu and Qiong Pan were responsible for the data analysis. Yuan Peng and Zhe Liang performed data interpretation and follow up. Yuefang Liu wrote the article. Qiong Pan revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Peng, Y., Liang, Z. et al. Parent-of-origin testing of prenatal copy number variations: a retrospective study of 167 family cases. Sci Rep 15, 5979 (2025). https://doi.org/10.1038/s41598-025-86487-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-86487-w
