
Abstract
Predicting drug-target interaction (DTI) stands as a pivotal and formidable challenge in pharmaceutical research. Many existing deep learning methods only learn the high-dimensional representation of ligands and targets on a small scale. However, it is difficult for the model to obtain the potential law of combining pockets or multiple binding sites on a large scale. To address this lacuna, we designed a large-kernel convolutional block for extracting large-scale sequence information and proposed a novel DTI prediction framework, named Rep-ConvDTI. The reparameterization method is introduced to help large-kernel convolutions capture small-scale information. We have also developed a gated attention mechanism to more efficiently characterize the interaction of drugs and targets. Extensive experiments demonstrate that Rep-ConvDTI achieves the most competitive performance against state-of-the-art baselines on the three benchmark datasets. Furthermore, we validated the potential of Rep-ConvDTI as a drug screening tool through model interpretative studies and drug screening experiments with cystathionine-β-synthase.
Similar content being viewed by others


Introduction
In the realm of medicinal chemistry, a pressing challenge is being confronted: despite the vast size of available compound database, only a minuscule fraction of these candidates show high affinity and specificity for binding to targeted proteins. Although there are many existing computational simulation methods1,2,3 for reducing the cost of drug development, finding a suitable lead compound still takes researchers months or even years4.
Recent years, we have witnessed a remarkable surge in artificial intelligence technology, notably with deep learning models excelling in diverse domains such as machine vision5, natural language processing6, and speech recognition7. Concurrently, the advent of high-throughput experimentation8 has led to a proliferation of biological data, enabling the development of virtual drug screening models powered by big data. By formalizing the virtual screening task into binary classification (DTI drug-target interaction) or regression (DTA drug-target affinity) tasks, many excellent deep learning modeling methods can be applied to solve drug screening problems. For example, DTINet9 formalizes the DTI task into the edge prediction problem of nodes in the graph. DrugVQA10 uses a method similar to the visual question-answering task, treating the target protein distance map as a picture and the ligand as a question, to obtain the prediction results.
One of the key challenges in current drug-target interaction prediction tasks is how to represent proteins and compounds as high-dimensional features that are both more informative and more discriminative. DeepDTA11 employs two distinct convolutional neural networks(CNNs) to encode amino acid sequences and SMILES(Simplified Molecular Input Line Entry System)12 strings, respectively, for making affinity predictions. DeepConv-DTI13 attempts to capture drug characterization with drug fingerprints14 and fully connected layers. To enrich the representations of compounds and proteins with more binding site information, DeepCDA15 combines long short term memory and convolutional neural networks to encode proteins and compounds and innovatively proposes a two-sided attention mechanism for encoding protein and compound substructures interaction strength. HyperAttentionDTI16 introduced a similar sequential attention mechanism, which assign an attention vector to each atom or amino acid. DrugMGR17 designed a GatedCNN-based Variational Autoencoder (VAE)18 structure to enhance the learning ability of CNNs for binding sites. DrugBAN19 present a deep bilinear attention network framework that capture pairwise local interactions between drug and protein though a bilinear interaction map and a bilinear pooling layer. The use of pre-trained models is considered an effective method for obtaining richer representations. FusionDTA20 first obtains protein representations through Transformer pre-training and then further trains an affinity prediction model. iNGNN-DTI21 combines Chemformer22 and ESM23 with graph data to characterize compounds and proteins. Besides, graph neural networks24,25,26,27 and multimodal28,29 data have been utilized to train more discriminative representations.
In summary, convolutional neural networks remain one of the primary methods for encoding proteins and compounds. Most of the existing deep learning prediction methods11,13,16, when encoding the primary structure of targets, obtain the global representation of the target from small local amino acid sequence information. Nevertheless, binding pockets often consist of discontinuous segments scattered along the protein’s peptide chain, or even spanning multiple chains. The reliance on small local sequences thus provides only a fragmented view of the binding site, failing to encompass its full complexity. As a result, these models struggle to accurately discern the holistic features of the target protein and are consequently unable to determine the potential law of binding the whole pocket to the ligand. In practice, a well-designed model should not only grasp the whole information of large-scale but also take into account the local small-scale binding sites information.
Recently, in the field of deep learning, using a few large convolutional kernels has been proven more effective than stacking numerous small kernels30,31, endowing models with larger effective receptive fields and stronger representation capabilities. Inspired by those work, we propose a large-kernel convolution-based drug and target interaction prediction model, Rep-ConvDTI. Specifically, a dilated reparameterization one-dimensional convolution was introduced in Rep-ConvDTI to help the model effectively extract sequence information of multiple target proteins and ligand compounds of different sizes by training multiple small convolution kernel in parallel while training large convolution weights. We also designed a gated attention mechanism to mine complex interactions between compound molecules and amino acids over a large range. In the end, The integrated learning method XGBoost32 was used to accurately decode the high-dimensional features and obtain the DTI prediction results.
In this paper, we present a detailed exposition of our drug-target interaction prediction model, Rep-ConvDTI, and substantiate its superior performance through comparative analysis against state-of-the-art methodologies in drug-target interaction prediction tasks. We apply Rep-ConvDTI to forecast interactions between cystathionine-β-synthase and a cohort of 22 drugs. The predicted outcomes validate the model’s reliability in real-world drug-target interaction prediction scenarios. The main contributions of this paper are twofold:
-
(1)
We introduce and design a dilated reparameterization 1D convolution method suitable for DTI prediction tasks.
-
(2)
We construct a DTI prediction framework capable of integrating both large-scale and small-scale information, and the results of visualization and wet experiments confirm its reliability.
Datasets
We selected DUD-E33, KIBA34, and Davis35 as the benchmark datasets to evaluate the performance of Rep-ConvDTI in the DTI prediction task.
The DUD-E dataset includes 102 targets and their affinities for 22,886 active compounds. For each active compound, 50 decoys with physico-chemical properties similar to those of the active compounds but inactive were used.
The Davis dataset contains bioactivity data on the interaction of 68 ligands with 442 targets, and the degree of binding between the target and the ligand is measured by the Kd value. We collected 30,056 combined data points and labelled the sample pairs with the Kd value of 10,000 nanomoles as negative samples and the others as positive samples.
The KIBA dataset contains 229 targets and 2116 ligands, in which each ligand and target has at least 10 sample pairs, and KIBA scores is used to measure the binding affinity between sample pairs. Similarly, we referred to previous works16,36 and marked the sample pairs whose KIBA scores are less than 12.1 as negative samples and the others as positive samples.
We adopted the method of random sampling10 to eliminate redundant negative samples and constructed a balanced dataset.The balanced datasets we constructed are summarized in Table 1. To systematically evaluate the acpability of Rep-ConvDTI in DTI prediction task, two different test methods are employed: “hot-start-for-protein” and “cold-start-for-protein”. The “hot-start-for-protein” method involves a training set that includes all the proteins present in the test set. In contrast, the “cold-start-for-protein” method uses a training set that does not contain any of the proteins from the test set.
Methods
The network architecture of Rep-ConvDTI
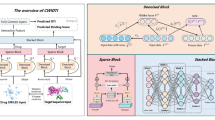
Our model framework is mainly composed of three parts: an input layer, a feature extraction layer, and a decoding layer. As shown in Fig. 1a, ligands and targets are first encoded into one-hot vectors, and the corresponding high-dimensional features are generated through the embedding layer. Then, the important features are extracted by the feature extraction layer through two different scales of convolutional neural networks, and finally, the decoding layer will produce the prediction results of the model. The details of each layer are detailed in the following sections.

(a) The network framework of the Rep-ConvDTI model. (b) The gated attention network architecture. (c) The calculation processes of the LGCNN block and SMCNN block include an SE block37 and layer normalization38. The only difference is that the LGCNN block utilizes a dilated reparam 1D conv in place of the ordinary one-dimensional convolution.
Input layer
Targets and ligands are represented in the form of amino acid sequences and SMILES strings in the dataset. We first transformed all SMILES into standardized SMILES, and canonical SMILES, and encoded each ligand into a one-hot representation by creating a corresponding character dictionary. Finally, the embedding layer converted these representations into embedding matrices, \(x^{D} = \left\{ {x_{1}^{D} ,x_{2}^{D} ,x_{3}^{D} \ldots ,x_{{L_{d} }}^{D} } \right\} \in R^{{V_{D} }}\). Similarly, each target can be represented as \(x^{P} = \left\{ {x_{1}^{P} ,x_{2}^{P} ,x_{3}^{P} \ldots ,x_{{L_{p} }}^{P} } \right\} \in R^{{V_{P} }}\) and, where \(\:{L}_{d}\) and \(\:{L}_{p}\) represent the length of the SMILES sequence and the amino acid sequence, respectively, and \(\:{V}_{D}\) and \(\:{V}_{P}\) are the dimensions of the ligand and target embedding features, respectively.
Feature extraction layer
LGCNN block
To effectively mine the latent information about a wide range of amino acid subsequences and ligand compound substructures, we designed a 1D convolution block, the LGCNN block, which is similar to the 2D convolution block commonly used in vision tasks, as shown in Fig. 1c. Given an input feature map \(\:x\in\:{R}^{L\times\:V}\), the LGCNN block applies a dilated reparam conv with a kernel size of \(\:k\) to distill low-dimensional features across \(\:C\) channels, yielding a transformed feature matrix \(\:{x}^{{\prime\:}}\in\:{R}^{L\times\:C}\). We set the filling quantity to \(\left\lfloor {k/2} \right\rfloor\) to ensure that the length of the feature matrix remains unchanged. After layer normalization, a squeeze-and-excitation block (SE block)37 is used to dynamically change the influence of features of different channels on the output to improve the contribution of channels containing associative information to the result. The calculation is shown as follows:
where \(\:\sigma\:\) and \(\:\delta\:\) are the sigmoid and ReLU activation functions, respectively; \(\:{W}_{1}\in\:{R}^{\frac{C}{r}\times\:c}\) and \(\:{W}_{2}\in\:{R}^{C\times\:\frac{c}{r}}\) are learnable weight parameters; and \(\:r\) is the dimension decrease ratio.
Dilated reparam 1D conv
Inspired by previous research30, we employed large-kernel one-dimensional convolution to extract high-dimensional features from targets and ligands and captured small-scale local information to help large-sized convolution during training. We then trained several-kernel convolution in parallel31, and their outputs were aggregated after layer normalization to obtain the final result. In this work, we found that the equivalent transformation method proposed in UniRepLKNet30, which used one nondilated small-kernel layer and multiple dilated small-kernel layers in parallel to enhance a nondilated large-kernel layer, is also helpful for improving the performance of one-dimensional large-kernel convolution. We defined a 1D convolution with a kernel size greater than or equal to 9 as a large-kernel convolution and maintain consistency in hyperparameters, including the large convolution kernel size \(\:K\), parallel convolution kernel size \(\:k\), and expansion rate \(\:r\), with the values used in30. As depicted in Fig. 2, when the kernel size K was set to 9, we trained four small kernel convolutions in parallel, with kernel sizes of 5, 3, 3, and 3. The dilation coefficient for each of these kernels was set to 1, 2, 3, and 4. Following the normalization layer, the outputs of all these layers were aggregated, effectively simulating a dense convolution kernel with a size of 9.

The structure and parameter perspective of the dilated reparam 1D conv when \(\:K=9\). From the parameter perspective, multiple dilated small-kernel layers can be equivalently transformed into a single large-kernel conv. Such a convolution kernel extension method is flexible, but it must be followed \(\:\left(k-1\right)r+1\le\:K\).
Gated attention
The gating mechanism, a well-established technique in recurrent neural networks39,40, has been proven effective in forgetting redundant information from previous time steps and updating the current information. This approach significantly mitigates the challenges of gradient vanishing and long-term memory loss in recurrent architectures. Based on this concept, we devised a gated attention mechanism that specifically discards information irrelevant to drug-ligand interaction during their characterization. Subsequently, this mechanism updates the corresponding eigenmatrix to facilitate the discovery of interaction regions within the local structure between the ligand and the target.
As shown in Fig. 1b, given ligand characteristic matrix \(X^{D} \in R^{{L_{D} \times C_{D} }}\) and target characteristic matrix \(X^{P} \in R^{{L_{P} \times C_{P} }}\), we first obtain their respective hidden layer representations through the fully connected layer:
where \(\:{W}_{D}\in\:{R}^{{C}_{D}\times\:C}\) and \(\:{W}_{P}\in\:{R}^{{C}_{P}\times\:C}\) are weight matrices. \(\:b\) is the bias vector. Then, the update gate is calculated by the following formula:
where \(\:{H}_{t}^{D}\in\:{R}^{{L}_{D}\times\:{L}_{P}\times\:C}\) and \(\:{H}_{t}^{P}\in\:{R}^{{L}_{D}\times\:{L}_{P}\times\:C}\) are the expanded dimensions of \(\:{h}_{t}^{D}\) and \(\:{h}_{t}^{P}\), respectively, \(\:F\) is the encoder-decoder network layer, \(\:\phi\:\) is the activation function \(\:Tanh\), and \(\:Mean(Input,\:dim)\) returns the result of averaging the dim dimensions. The updated ligand and target characteristics can be expressed as:
where \(\:\alpha\:\) is the hyperparameter. Then, \(\:{X}_{update}^{D}\) and \(\:{X}_{update}^{P}\) are fed into the global maximum pooling layer and are concatenated into a feature vector. We use the fully connected layer to convert this vector into a hybrid feature \(\:{v}_{f}\) as the input feature of the decoding layer.
Decoding layer
In this study, to correctly decode the hidden layer characteristics of complex drugs and target mixing and improve the accuracy of model prediction, we used the ensemble learning model XGBoost to output the final interaction prediction results. The advantages of XGBoost are shown as follows: (1) Compared with a multilayer perceptron, ensemble learning is better interpretable by training multiple weak classifiers to build complex nonlinear relationships among features. (2) Compared with other ensemble learning methods, XGBoost can choose a decision tree or linear classifier as the base classifier and introduce regular terms into the objective function to improve the prediction performance and generalizability of the model.
For XGBoost, the prediction result of the input feature \(\:{v}_{f}\) of the sample against \(\:i\) in the \(\:k\)th iteration is as follows:
where \(\:{\widehat{y}}_{i}^{(t-1)}\) is the prediction result of the \(\:k\)-1 tree and \(\:{f}_{t}\left({v}_{f}\right)\) is the output result of the \(\:k\)th tree.
Implementation and evaluation strategy
Performance Metrics: Our evaluation encompasses four quintessential metrics for assessing binary classification tasks, namely Accuracy, Precision, Recall, the Area Under the Receiver Operating Characteristic Curve (AUROC) and the Area Under the Precision-Recall Curve (AUPRC), collectively serving as pivotal indicators of model performance.
Training: We built the Rep-ConvDTI model using the PyTorch 2.0 framework and first replaced the decoding layer with a multilayer perceptron, set the size of each training batch to 128, and trained 100 cycles on an NVIDIA A800 GPU using the AdamW optimizer41 with a learning rate of 0.0001. Then, the parameters of the input layer and the feature extraction layer are fixed, and the output results of the feature extraction layer are used as inputs to train the XGBoost model.
To show the importance of the selected architecture, we report the obtained results for XGBoost and Rep-ConvDTIMLP. For XGBoost, The one-hot encoded feature vectors of the drug and target are directly used as input to XGBoost without undergoing neural network encoding, and then the predicted results are obtained for comparative experiments. For Rep-ConvDTIMLP, the decoding layer is replaced with a fully connected neural network for comparative experiments. To evaluate the performance of our proposed Rep-ConvDTI method for DTI prediction, we compare our model with three baselines of DTI prediction methods: DeepDTA11, DeepConv-DTI13 and HyperAttentionDTI16. For those models, We utilized their open-source code and trained the model using the parameters provided in their paper for comparative experiments.
Results
Performance evaluation with “hot-start-for-protein”
In “hot-start-for-protein” case, we adopted 5-fold cross-validation for Davis and KIBA dataset and set the ratio of the training, validation, and testing to 16:4:5. According to the experimental results in Table 2, our model shows the best performance in terms of the five indices. On the DAVIS dataset, the performance of Rep-ConvDTI is significantly better than that of XGBoost, showing an improvement of approximately 5.9%. This demonstrates the substantial encoding power and effectiveness of the neural network. Additionally, compared to three other prediction methods based on convolutional neural networks, Rep-ConvDTIMLP achieved improvements of 0.008, 0.018, 0.011 and 0.003 in terms of accuracy, recall, AUROC and AUPRC, respectively. In particular, when compared with HyperAttentionDTI, which features a similar attention-mechanism module, Rep-ConvDTIMLP achieved a significant 5.7% increase in recall. This demonstrates that the dilated reparameterize convolution we use possess superior feature learning capabilities compared to ordinary convolutional networks. Finally, in comparison with Rep-ConvDTIMLP, which uses a fully connected layer for decoding, the complete Rep-ConvDTI outperforms it across all five metrics, indicating that XGBoost has better decoding capabilities than the fully connected neural network. On the KIBA dataset, we observed similar results, our approach achieves 3.93%, 4.15%, 2.98%, and 3.21% improvements in accuracy, recall, AUROC and AUPRC over the baseline model’s best performance. Notably, HyperAttentionDTI exhibited a 10.4% lower recall rate compared to DeepDTA, while the decrease was only 3.3% in the DAVIS dataset. This discrepancy may be attributed to the KIBA dataset’s large diversity of ligands, which causes the attention blocks in HyperAttentionDTI to bias towards predicting non-interactions when handling a vast number of ligands. In contrast, the dilated reparameterize convolution and gated attention mechanism we employed can effectively mitigate this phenomenon.
The “hot-start-for-protein” experimental results on the DAVIS and KIBA benchmark datasets indicate that the gated attention mechanism enhances model performance, particularly in precision, although it has some impact on recall. However, this drawback is mitigated by the dilated reparameterized convolution. Additionally, the XGBoost decoder reduces potential prediction biases during training, leading to more reliable prediction outcomes.
Performance evaluation with “cold-start-for-protein”
The discovery of new targets plays a crucial role in drug development but also presents a significant challenge for virtual drug screening. To verify the predictive ability of our model in the absence of prior knowledge about new targets, we adopted 5-fold cross-validation for DUD-E dataset, we used 5-fold cross-validation for the DUD-E dataset. Unlike the hot-start case, we maintained a 4:1 protein ratio between the training and test sets, and a fifth of the training set was randomly selected as the validation set. Figure 3 shows the experimental results, demonstrating that our approach outperforms all baselines in terms of Recall, AUROC and AUPRC. On average, Rep-ConvDTI performs better by 18.84%, 4.02%, 7.3%, 5.53% and 3.8% than XGBoost, DeepDTA, DeepConv-DTI, HyperAttentionDTI and Rep-ConvDTIMLP. The figure shows that HyperAttention exhibits the largest fluctuation in recall rate during five-fold cross-validation, with a standard deviation of 0.1. In contrast, Rep-ConvDTIMLP demonstrates significantly greater stability in recall rate. Moreover, Rep-ConvDTI shows notable improvements over Rep-ConvDTIMLP in both AUROC and AUPRC, further corroborating the conclusions drawn from our “hot-start-for-protein” experiments.

Comparison result of DUD-E dataset with “cold-start-for-protein”.
Ablation study
In this study, The Dilated Reparam 1D Conv was used to extract ligand and target features, and a novel gated attention mechanism was designed to explore the complex interaction between compound molecules and amino acid side chains. Here, we designed three variants of Rep-ConvDTI for ablation experiments to verify the validity of each module in the model. These included Variant-1, 2, and 3. In Variant-1, we remove the compression and extraction layers in each convolutional block to ensure that the features on each channel have the same weight and verify the effect of SE blocks on model performance. For Variant-2, we delete all the reparameterization steps of large-kernel blocks, abandon all the small-kernel layers trained in parallel, and turn all the dilated convolution blocks back into ordinary convolutional networks; then, we verify the influence of reparameterization methods on network performance. In Variant-3, to verify the effect of the gated attention layer on model performance, high-dimensional features of the target and ligand are directly spliced into the decoding layer, and the features of the target and ligand do not interact. Table 3 shows the performances of the different variants on the Davis dataset.
As seen from Table 3, each variant has a different degree of performance degradation compared to the unabridged Rep-ConvDTI model. Specifically, the performance of Variant-2 showed a significant decrease of approximately 3.2%, which indicates that parallel multiple small-kernel layers can effectively help large-kernel convolution to extract features, and structural reparameterization is an effective way to improve the performance of large convolution kernels. In addition, the AUROC and AUPRC in the benchmark dataset of Variant-3 are reduced by an average of 0.013, which also proved that the gated attention mechanism can help the model find the key sites involved in the binding of drugs and targets and is an indispensable part of the model when mining interaction information. Finally, experiments for Variant-1 show that dynamic weighting of feature channels can effectively improve model performance when feature extraction is carried out in convolutional networks.
Further, we conducted detailed experiments (the results are available in Supplementary) on hyperparameters such as the number of convolutional layers, kernel size (shown in Supplementary Table S1), and the number of small kernels used in parallel training (shown in Supplementary Table S2). By systematically tuning these hyperparameters, we not only optimized model performance but also uncovered the specific impacts of different settings on model behavior.
Interpretability analysis
Here, we further analyze the interpretability of the model by visualizing the weight parameters of the gated attention layer. From the crystal structure provided by the DUD-E database, we selected two samples that were considered by the model to have a high probability of binding in the test process for analysis, namely, the crystal structure of protein kinase C beta-type (kpcb) binding with Q27464378 and the crystal structure of mitogen-activated protein kinase 10 (mk10) binding with 2zdt. First, we obtained a hidden layer representation of the input target and ligand and then calculated the updated amount of the update gate for each molecule and projected the value into 0–1 using a standardized method. The final results are shown in Fig. 4.

Weight visualization of Rep-ConvDTI for the kpcb crystal structure (a) and the mk10 crystal structure (b). Amino acids with high attention weights given by Rep-ConvDTI are marked in red. The yellow area shows the shape of the protein-binding pocket. We highlighted the binding sites that Rep-ConvDTI predicted correctly.
As shown in Fig. 4, Rep-ConvDTI has identified specific amino acids involved in ligand binding for two protein targets: kpcb and mk10. For kpcb, Val423 was found to bind to ligands, while Asn424 and Gly425 formed binding pockets. Similarly, for the mitogen-activated protein mk10, our method accurately pinpointed Leu148 and Asn194 as two amino acid subunits located at the binding site. However, for binding ligands, it is difficult for us to observe obvious binding information from the attention perspective given by Rep-ConvDTI, which indicates that Rep-ConvDTI framework we designed can provide a reference for potential binding regions, but further improvement and perfection are needed to achieve full reliability.
In-lab validation
To verify the predictive reliability of our model in a real-world laboratory, we performed virtual drug screening for cystathionine-β-synthase (CBS). CBS, the first (and rate-limiting) enzyme in the transsulfuration pathway, is involved in the metabolism of homocysteine (a cytotoxic molecule and cardiovascular risk factor) and the generation of hydrogen sulfide (H2S) and is an important mammalian enzyme in health and disease. Preclinical data show that the inhibition or inactivation of CBS has beneficial effects on the treatment of some patients with Down syndrome and various cancers42. We first used docking to screen 22 active ligands with a high probability of binding to CBS targets from ZINC1543. After that, Rep-ConvDTI was used for further screening, ligands with binding probabilities greater than 50% of the model output were regarded as binding ligands, and other ligands were regarded as unbinding ligands. In Table 4, we list the comparison between the model prediction results and the experimental results for 22 ligands. We found that many of the predicted results of Rep-ConvDTI were identical to the experimental results, which indicates the potential of Rep-ConvDTI as a drug screening tool and is expected to help researchers select promising active ligands.
Discussion and conclusion
In this paper, we designed a new DTI prediction model based on a dilated reparameterize convolution network, Rep-ConvDTI. The experimental results on three benchmark datasets show that our model has the best performance under 5-fold cross-validation compared to other advanced prediction approaches. Then, we verified the influence of each module in the model on the model performance in the ablation experiment. Moreover, we conducted experiments on the kpcb and mk10 targets to evaluate the model’s interpretability, demonstrating that the gated attention mechanism can help the model uncover valuable interaction information. Finally, we experimentally validated the binding interactions of 3 candidate compounds with the CBS target and excluded 15 compounds that Docker predicted to have high binding potential. This demonstrates the potential of Rep-ConvDTI as an auxiliary or even a replacement tool for Docker in virtual drug screening.
The outstanding performance of Rep-ConvDTI can be attributed to three key aspects. (i) Application of reparameterized convolutions: we utilized 1D reparameterized convolutions instead of traditional CNNs. This approach allows the convolution kernels to have larger sizes while focusing on small-scale local information, significantly enhancing model performance, especially in terms of recall. This improvement provides new insights for other CNN-based drug-target interaction prediction models. (ii) Desgin of gated attention: compared to HyperAttentionDTI, our gated attention mechanism not only improves model interpretability but also delivers better performance. (iii) Introduction of XGBoost: We introduced XGBoost as a decoder, which further optimized the model’s predictive capabilities by reducing prediction biases during training and making the prediction results more stable and reliable.
This work has numerous potential biological applications in predicting the interaction sites with small molecules from the simplest source of information, the amino acid sequence. For example, small-molecule binding site prediction can be attempted for a large number of artificially designed novel fusion protein drugs or protein tools, and these large numbers of artificially designed proteins are often so numerous that it is difficult to realize the acquisition of accurate 3D structures in batch. Another example, as we all know, is that in addition to the catalytic pockets or known binding pockets, there are often one or more other allosteric pockets on the surface of a protein, and drug design targeting these important allosteric pockets can often achieve unique advantages such as high selective activity or high activity. Using our present research method, it is possible to try to utilize the simple primary structural sequence information of the target protein to predict the potential allosteric pocket on its surface, which is a nice complement to the existing software for predicting allosteric pockets based on the 3D structure.
Last but not least, although Rep-ConvDTI provides a powerful deep learning tool for drug-target interaction prediction, there are certain limitations to our model. Our model achieved the best performance in comparative experiments, but the introduction of reparameterized convolutions and gated attention significantly increased the number of model parameters, leading to higher costs for training and fine-tuning. We aim is to develop a coding modality suitable for DTI prediction, where the approximate area of a potential binding site or pocket can be inferred using the primary structure of the protein alone, providing effective support for drug developers, but the current gated attention mechanism has not yet fully achieved this goal. These issues warrant further investigation and improvement in our future work.
Data availability
All data used in this paper are publicly available and can be accessed at https://staff.cs.utu.fi/~aatapa/data/DrugTarget/ for the Davis dataset, http://dude.docking.org for the DUD-E dataset, http://github.com/hkmztrk/DeepDTA for the KIBA dataset.
Code availability
The codes of Rep-ConvDTI are available at http://github.com/DMP321/Rep-ConvDTI.
References
Ewing, T. J. A., Makino, S., Skillman, A. G. & Kuntz, I. D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 15, 411–428 (2001).
Thomsen, R. & Christensen, M. H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 49, 3315–3321 (2006).
Kuntz, I. D., Blaney, J. M., Oatley, S. J., Langridge, R. & Ferrin, T. E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 161, 269–288 (1982).
Xia, X., Zhu, C., Zhong, F. & Liu, L. MDTips: A multimodal-data-based drug–target interaction prediction system fusing knowledge, gene expression profile, and structural data. Bioinformatics 39, btad411 (2023).
Liu, Z. et al. Swin Transformer. arXiv.org. https://arxiv.org/abs/2103.14030v2 (2021).
Vaswani, A. et al. Attention is all you need. https://doi.org/10.48550/arXiv.1706.03762 (2017).
Dong, L., Xu, S., Xu, B. & Speech-transformer a no-recurrence sequence-to-sequence model for speech recognition. In IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP) 5884–5888 (IEEE, 2018). https://doi.org/10.1109/ICASSP.2018.8462506.
Dunlop, J., Bowlby, M., Peri, R., Vasilyev, D. & Arias, R. High-throughput electrophysiology: An emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 7, 358–368 (2008).
Luo, Y. et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 8, 573 (2017).
Zheng, S., Li, Y., Chen, S., Xu, J. & Yang, Y. Predicting drug–protein interaction using quasi-visual question answering system. Nat. Mach. Intell. 2, 134–140 (2020).
Öztürk, H., Özgür, A. & Ozkirimli, E. DeepDTA: Deep drug–target binding affinity prediction. Bioinformatics 34, i821–i829 (2018).
Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Lee, I., Keum, J. & Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLOS Comput. Biol. 15, e1007129 (2019).
Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754 (2010).
Abbasi, K. et al. DeepCDA: Deep cross-___domain compound–protein affinity prediction through LSTM and convolutional neural networks. Bioinformatics 36, 4633–4642 (2020).
Zhao, Q., Zhao, H., Zheng, K. & Wang, J. HyperAttentionDTI: Improving drug–protein interaction prediction by sequence-based deep learning with attention mechanism. Bioinformatics 38, 655–662 (2022).
Li, X. et al. DrugMGR: A deep bioactive molecule binding method to identify compounds targeting proteins (2024).
Kingma, D. P. & Welling, M. Auto-encoding variational bayes. https://doi.org/10.48550/ARXIV.1312.6114 (2013).
Bai, P., Miljković, F., John, B. & Lu, H. Interpretable bilinear attention network with ___domain adaptation improves drug–target prediction. Nat. Mach. Intell. 5, 126–136 (2023).
Yuan, W., Chen, G. & Chen, C. Y. C. FusionDTA: Attention-based feature polymerizer and knowledge distillation for drug-target binding affinity prediction. Brief. Bioinform. 23, bbab506 (2022).
Sun, Y., Li, Y. Y., Leung, C. K. & Hu, P. iNGNN-DTI: Prediction of drug–target interaction with interpretable nested graph neural network and pretrained molecule models. Bioinformatics 40, btae135 (2024).
Irwin, R., Dimitriadis, S., He, J. & Bjerrum, E. Chemformer A pre-trained transformer for computational chemistry. https://doi.org/10.26434/chemrxiv-2021-v2pnn (2021).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. 118, e2016239118 (2021).
Nguyen, T. et al. GraphDTA: Predicting drug–target binding affinity with graph neural networks. Bioinformatics 37, 1140–1147 (2021).
Yang, Z. MGraphDTA: Deep multiscale graph neural network for explainable drug–target binding affinity prediction. Chem. Sci. (2022).
Yazdani-Jahromi, M. et al. AttentionSiteDTI: An interpretable graph-based model for drug-target interaction prediction using NLP sentence-level relation classification. Brief. Bioinform. 23, bbac272 (2022).
Wang, Y. et al. ZeroBind: A protein-specific zero-shot predictor with subgraph matching for drug-target interactions. Nat. Commun. 14, 7861 (2023).
Rafiei, F. et al. DeepTraSynergy: Drug combinations using multimodal deep learning with transformers. Bioinformatics 39, btad438 (2023).
Dehghan, A., Razzaghi, P., Abbasi, K., Gharaghani, S. & TripletMultiDTI Multimodal representation learning in drug-target interaction prediction with triplet loss function. Expert Syst. Appl. 232, 120754 (2023).
Ding, X. et al. UniRepLKNet: A universal perception large-kernel ConvNet for audio, video, point cloud, time-series and image recognition. https://doi.org/10.48550/arXiv.2311.15599 (2023).
Ding, X. et al. Scaling up your kernels to 31 × 31: Revisiting large kernel design in CNNs. http://arxiv.org/abs/2203.06717 (2022).
Chen, T., Guestrin, C. & XGBoost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 785–794 (ACM, 2016). https://doi.org/10.1145/2939672.2939785.
Mysinger, M. M., Carchia, M., Irwin, J. J. & Shoichet, B. K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6582–6594 (2012).
Tang, J. et al. Making sense of Large-Scale Kinase Inhibitor Bioactivity Data sets: A comparative and integrative analysis. J. Chem. Inf. Model. 54 (2014).
Davis, M. I. et al. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 (2011).
He, T., Heidemeyer, M., Ban, F., Cherkasov, A. & Ester, M. SimBoost: A read-across approach for predicting drug–target binding affinities using gradient boosting machines. J. Cheminform. 9, 24 (2017).
Hu, J., Shen, L., Albanie, S., Sun, G. & Wu, E. Squeeze-and-excitation networks. http://arxiv.org/abs/1709.01507 (2019).
Ba, J. L., Kiros, J. R. & Hinton, G. E. Layer normalization. https://doi.org/10.48550/arXiv.1607.06450 (2016).
Hochreiter, S. & Schmidhuber, J. Long short-term memory. Neural Comput. 9, 1735–1780 (1997).
Cho, K. et al. Learning phrase representations using RNN encoder-decoder for statistical machine translation. https://doi.org/10.48550/arXiv.1406.1078 (2014).
Loshchilov, I. & Hutter, F. Decoupled weight decay regularization. arXiv.org. https://arxiv.longhoe.net/abs/1711.05101v3 (2017).
Zuhra, K., Augsburger, F., Majtan, T. & Szabo, C. Cystathionine-β-synthase: Molecular regulation and pharmacological inhibition. Biomolecules 10, 697 (2020).
Sterling, T. & Irwin, J. J. ZINC 15—ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Acknowledgements
This works was supported by the General Project of Basic Research Plan of Shenyang Institute of Automation, Chinese Academy of Sciences in 2023 (2023JC2K03) , the Science and Technology Plan for Artificial Intelligence of Liaoning Province (2023JH26/10300014) and the Nature Foundation (Basic Research) Special Project of Shenyang (22-315-6-20).
Author information
Authors and Affiliations
Contributions
M.D. was responsible for Rep-ConvDTI model design and paper writing as well as drawing all the figures in the paper. J.W. provided multiple baseline models and conducted detailed comparison tests and ablation experiments. Yiming Zhao and Yongjia Zhao collected data from the Davis, KIBA, and DUD-E datasets and processed them. H.C. provided CBS in-lab data on CBS target and gave guidance in the field of pharmacy within the discussion section of the article. Z.W. contributed various ideas and comments to improve Rep-ConvDTI. All authors read the approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Deng, M., Wang, J., Zhao, Y. et al. Predicting drug and target interaction with dilated reparameterize convolution. Sci Rep 15, 2579 (2025). https://doi.org/10.1038/s41598-025-86918-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-86918-8
