
Abstract
Extracellular vesicles (EVs) are important paracrine mediators derived from various cells and biological fluids, including plasma, that are capable of inducing regenerative effects by transferring bioactive molecules such as microRNAs (miRNAs). This study investigated the effect of mesenchymal stem cell-derived extracellular vesicles (MSC-EVs) isolated from umbilical cord blood and human umbilical cord plasma-derived extracellular vesicles (UCB-EVs) on wound healing and scar formation reduction. Spatial transcriptomics (ST) was used to study the effects of MSC-EVs and UCB-EVs on the heterogeneity of major cell types and wound healing pathways in mouse skin tissue. MSC-EVs and UCB-EVs were isolated using ultracentrifugation and identified using transmission electron microscopy, nanoparticle tracking analysis, and western blot. The effects of MSC-EVs and UCB-EVs on human dermal fibroblast-adult cell (HDF-a) migration and proliferation were evaluated using cell scratch assays, cell migration assays, and cell proliferation assays. In vivo, MSC-EVs and UCB-EVs were injected around full-cut wounds to evaluate their efficacy of wound healing by measuring wound closure rates and scar width and performing histological analysis. ST was performed on skin tissue samples from mice in each group after wound healing to analyze the heterogeneity of major cell types compared with the control group and investigate potential mechanisms affecting wound healing and scar formation. In vitro experiments demonstrated that MSC-EVs and UCB-EVs promoted the proliferation and migration of HDF-a cells. Local injection of MSC-EVs and UCB-EVs into the periphery of a mouse skin wound accelerated re-epithelialization, promoted wound healing, and reduced scar width. ST analysis of skin tissue from each group after wound healing revealed that MSC-EVs and UCB-EVs reduced the relative expression of marker genes in myofibroblasts, regulated wound healing, and decreased scar formation by reducing the expression of the TGF-β signaling pathway and increasing the expression of the Wnt signaling pathway. The results suggest that MSC-EVs and UCB-EVs play a significant role in the activity of cord blood plasma-derived mesenchymal stem cells and cord blood plasma. They can be considered promising new agents for promoting skin wound healing.
Similar content being viewed by others

Background
The skin, the body’s largest organ, is in direct contact with the external environment, which not only protects the body from the invasion of external pathogens but also has functions such as in sweating and sensation. Skin tissue damage can be caused by mechanical, physical, chemical, and biological factors, and the process of repairing this damage is called tissue repair. In certain pathological conditions, the typical process of wound healing is disrupted and prolonged, resulting in chronic non-healing wounds, such as diabetic ulcers or pathological scars1,2. Scar formation is a common outcome of skin injury during wound healing in adults, and severe cases can lead to psychological disorders and physical deformities3,4,5. Current methods for promoting wound healing and reducing scarring include skin grafting, laser therapy, and topical application of certain growth factors or histiocytes6,7,8,9. However, these methods can result in atrophic scarring, abnormal pigmentation, skin necrosis, and other adverse consequences10. Therefore, it is important to find a stable, effective, and safe method to shorten the healing time of skin tissue trauma and inhibit scar formation.
Extracellular vesicles (EVs) are present in almost all biological fluids11and can also be released from various cell types12. As subcellular components secreted by cells, EVs contain a variety of biomolecules, such as proteins, miRNAs, and lipids13, which can mediate intercellular communication by initiating a series of reactions in recipient cells and exert biological effects as paracrine factors of stem cells. EVs have been studied in the field of skin injury repair and regeneration as they can promote the repair and regeneration of lost skin tissue by promoting the proliferation and migration of fibroblasts (FBs) and angiogenesis, regulating inflammatory response and extracellular matrix (ECM) remodeling, and reducing scar formation. For example, Exosomes (Exos) derived from endothelial progenitor cells (EPCs) promote angiogenesis through the Erk1/2 signaling pathway, thereby accelerating skin wound healing14. EVs secreted by stem cells derived from cord blood can promote skin cell proliferation, migration, and angiogenesis to promote wound healing15. Epidermal stem cell-derived Exos (EPSC-Exos) can play an important role in inhibiting myofibroblasts (MFBs) differentiation by reducing the expression of TGF-β1 in dermal FBs16. EVs derived from human amniotic fluid stem cells (hAFSCs) can accelerate wound healing; promote regeneration of hair follicles, nerves, and blood vessels; and enhance the proliferation of skin cells and the natural distribution of collagen during wound healing17. These findings suggest that Exos-based therapy is a promising method for promoting wound healing.
Spatial transcriptomics (ST) is a technique for analyzing and describing the expression profile of specific cell types in spatial dimensions, which can address the transcriptional state of tissues in different spatial locations. It is a powerful tool for mapping cell-cell interactions and cell diversity in tissues. In dermatology, ST has been applied to mouse wound models at different time points after trauma to establish the spatial transcriptomic map of FBs populations during wound healing, revealing the potential mechanisms controlling FBs fate during migration, proliferation, and differentiation after skin injury and re-examining the typical stages of wound healing18.
In this study, we isolated EVs (MSC-EVs and UCB-EVs) from cord blood-derived mesenchymal stem cell culture medium and cord blood plasma and investigated their effects on skin wound healing and scar formation at the cellular and animal levels. In addition, ST sequencing was performed on mouse skin samples treated with MSC-EVs and UCB-EVs to understand the relative expression levels of myoblast marker genes in tissue samples and expression of related pathways during wound healing. This further revealed the effectiveness of MSC-EVs and UCB-EVs in promoting wound healing and reducing scar formation at the molecular level.
Methods and materials
Cells and animals
Mesenchymal stem cells culture supernatant and cord blood plasma were provided by Henan Yinfeng Bioengineering Technology Co., LTD. HDF-a cells were purchased from Otwo Biotech. SPF grade male BALB/c mice aged 6–8 weeks (26–30 g) purchased fromLiaoning Changsheng Biotechnology Co., LTD.
Isolation and purification of MSC-EVs and UCB-EVs
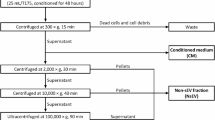
Cell culture supernatant19 or plasma20 diluted 1:1 with PBS was centrifuged at low speed (4 °C, 2000×g, 30 min; 4 °C, 10,000×g, 45 min). The suspensions were filtered through a 0.22 μm debacterialization filter. Ultracentrifugation (Beckman) was performed twice more (4 °C, 110,000×g, 70 min). The supernatant was discarded, and the precipitate was resuspended in 1.5-2 mL PBS, transferred into a 2 mL EP tube, and preserved at -80 °C.
Identification of MSC-EVs and UCB-EVs
Transmission electron microscopy (TEM) and nanoparticle tracking analysis (NTA) were conducted at Guangzhou Huayinkang Company.
The western blot (WB) process was as follows16: protein and 5× SDS loading buffers were mixed at a 4:1 ratio, and the protein was denatured (99 °C, 10 min). A 10% gel was prepared for electrophoresis. After completion, PVDF membrane transfer (Millipore) was performed. The membrane was blocked with 5% BSA at room temperature for 2 h. Primary antibodies (mouse anti-CD63 (Abcam), mouse anti-TSG101 (Abcam)) were incubated at 4 °C for 12–16 h. The membrane was washed and incubated with a secondary antibody (sheep anti-mouse (Proteintech) ) for 2 h. The expression of CD63 and TSG101 was measured using a GE imaging system.
Fluorescence tracing of MSC-EVs and UCB-EVs
A total of 3 × 104 HDF-a cells per well were prepared in a 24-well plate. EVs was mixed with PBS and PKH67 fluorescent dye (Sigma-Aldrich) in a 25:250:1 ratio and incubated at 37 °C for 5 min. One milliliter of PBS containing 0.5% BSA was added to terminate the labeling reaction. The labeled EVs were re-enriched by ultracentrifugation (120,000×g, 4°C, 60 min) and resuspended in PBS. Labeled EVs at 80 µg/mL were added to 24-well plates and cultured for 24 h, followed by fixation with 4% paraformaldehyde for 20 min. DAPI (Sigma-Aldrich) staining was performed for 1 h21. The cells were covered on a slide with an anti-fluorescence quencher and observed under a fluorescence microscope (Leica).
Culture of HDF-a
HDF-a cells were cultured in RPMI-1640 (CORNING) medium containing 10% FBS (37 °C, 5% CO2). When the cells were 90% confluent, the supernatant was discarded, and the cells were washed 1–2 times with D-PBS (CORNING). Subsequently, 1 mL of 0.25% EDTA pancreatic enzyme (CORNING) was added and digested at 37 °C for 2 min. The digestion was terminated with 1 mL of RPMI-1640 medium containing 10% FBS (Oricell) and centrifuged at 1000 rpm for 4 min. The cells were inoculated into a culture dish and placed in an incubator (SANYO) (37 °C, 5% CO2) for further culture.
Cell scratch assay
A straight line was drawn on the back of each well in a six-well plate at 1-cm intervals. A total of 5 × 105 HDF-a cells were added to each well (n = 3). Once the cells reached confluence, scratches were made with a 200 µL pipette tip perpendicular to the well and line. The wells were then washed with D-PBS, and serum-free medium was added. Three groups were established: control (100 µL/mL D-PBS), MSC-EVs (100 µg/mL), and UCB-EVs (100 µg/mL). The plates were cultured at 37 °C in a 5% CO2 incubator for 0, 12, and 24 h. ImageJ software was used to measure the area and average width of the scratches to calculate the migration rate. The closed area of the scratch was calculated as follows: migration area (%)=(A0-An)/A0 × 100, where A0 is the initial scratch area and An is the remaining area.
Cell migration assay
In a low-serum (1% FBS) medium with or without MSC-EVs (100 µg/mL) and UCB-EVs (100 µg/mL), 1 × 104 HDF-a cells per well (n = 3) were inoculated into the upper chamber of a 24-well plate containing an 8-µm pore filter. Complete culture medium without EVs (containing 10% FBS) was added to the lower chamber. After 24 h, the cells attached to the upper surface of the filter were removed. The migrated HDF-a cells were fixed with 4% paraformaldehyde for 30 min and stained with 0.5% crystal violet for 30 min. The number of migrated cells was observed under an optical microscope.
Cell proliferation (CCK-8) assay
A total of 5 × 103 HDF-a cells per well (n = 6) were inoculated into 96-well plates and cultured in a medium containing MSC-EVs and UCB-EVs at different concentrations (80, 100, 120 µg/mL). A cell-free group served as the blank control. At 12, 24, 48, and 72 h, CCK-8 solution (Solarbio) (10 µL) was added to each well and incubated at 37 °C for 3 h. Absorbance was measured at 450 nm using an enzyme-labeled instrument (Billerica). Cell viability was calculated as follows:
\(\:\text{C}\text{e}\text{l}\text{l}\:\text{v}\text{i}\text{a}\text{b}\text{i}\text{l}\text{i}\text{t}\text{y}=\frac{\text{d}\text{o}\text{s}\text{i}\text{n}\text{g}\:\text{g}\text{r}\text{o}\text{u}\text{p}\:\text{O}\text{D}-\text{b}\text{l}\text{a}\text{n}\text{k}\:\text{g}\text{r}\text{o}\text{u}\text{p}\:\text{O}\text{D}}{\text{c}\text{o}\text{n}\text{t}\text{r}\text{o}\text{l}\:\text{g}\text{r}\text{o}\text{u}\text{p}\:\text{O}\text{D}-\text{b}\text{l}\text{a}\text{n}\text{k}\:\text{g}\text{r}\text{o}\text{u}\text{p}\:\text{O}\text{D}}\)×100%
Quantitative real-time PCR (qRT-PCR)
HDF-a cells were inoculated in six-well plates and cultured for a period of time. The medium was then replaced with EVs-free medium, and MSC-EVs, UCB-EVs (100 µg/mL), and PBS were added (n = 3). After 24 h, RNA was extracted from HDF-a cells using the Trizol (Beyotime) method. For mouse skin tissue, 0.1 g was placed into a grinding tube, and tissue RNA was extracted using the Trizol method. According to the instructions of the Prime Script™ RT reagent kit, RNA samples were reverse-transcribed and amplified into cDNA by PCR. The expression of related genes was analyzed using qRT-PCR. GAPDH was used as the reference gene, and target gene expression was calculated using the comparative cycle threshold method (2−ΔΔCt method). The primer sequences are shown in Table 1.
Mouse skin wound model and treatment
A skin wound model was established in SPF grade male BALB/c mice aged 6–8 weeks (26–30 g). The back of each mouse was shaved and waxed. After 4–5 h, mice were induced to complete anesthesia with 2-2.5% isoflurane and maintained with 1-1.5% isoflurane to create a full-cut skin wound (10 mm in diameter)20,22. Forty-five mice were randomly divided into 3 treatment groups (n = 15). Starting from day 3, MSC-EVs, UCB-EVs, or PBS was injected subcutaneously around the wound for 5 consecutive days (40 µg of EVs diluted in 200 µL PBS per day). The wounds were photographed during the healing process. All wounds were measured using calipers, and the area of each wound was evaluated using Image Pro Plus 6 software on days 3, 7, 11, 15, and 17. Wound healing rate was calculated as (%)=(A0-At)/A0 × 100, where A0 is the initial wound area and At is the wound area on postoperative days 3, 7, 11, 15, and 17. On the 17th day, mice were euthanized using cervical dislocation method, and wound skin samples were collected from each treatment group. This investigation was carried out in compliance with the ARRIVE criteria for reporting research involving animals, guaranteeing comprehensive documentation of the experimental design, techniques, and results. The Animal Research Committee of Henan Agricultural University granted approval for the experimental protocol. (Ref. No.: HNND2024092901). Notably, all procedures involving animals were conducted in accordance with relevant guidelines and regulations, ensuring compliance with ethical standards for the care and use of laboratory animals.
Histopathological analysis
Skin samples from each treatment group were collected and immediately placed into 50 mL centrifuge tubes containing 4% paraformaldehyde ( Biosharp) for fixation. After rinsing and dehydrating with a series of different concentrations of alcohol, the samples were embedded in paraffin. HE staining was performed to observe histological changes, new epidermis, dermis, regenerated hair follicles, and adipocytes under an optical microscope. The scar width was measured, and the percentage of new epithelium was evaluated14. The percentage of re-epithelialization (E%) was calculated as: E%=(WN/WO)×100, where WO is the original wound area and WN is the length of newly formed epithelial cells across the wound surface. Masson’s trichrome staining was performed, and photographs were taken under the optical microscope to observe the degree of collagen maturity and condition of wavy fibers in the wound.
ST analysis
The sample was processed as follows: Skin samples (6.5 × 6.5 mm², thickness > 3 mm) were taken from mice in different treatment groups on day 17. OCT embedding agent (SAKURA) was pre-cooled on crushed ice for 30 min, and an appropriately sized embedding mold was selected to embed the skin tissue. Frozen Sects. (5–10 μm) were prepared at -20 °C. The fixed samples were stained with HE and photographed to determine histological information.
The tissue was permeabilized, intracellular mRNA was released, and a probe with spatial barcode information was bound. cDNA was synthesized on the slide, samples were digested, the probe was recovered, and cDNA was amplified. The library was constructed and sequenced using an Illumina high-throughput sequencing instrument to obtain sequencing data.
Statistical analysis
All tests were performed with at least 3 replicates per group, and in vitro tests were repeated at least 3 times. Data from each group of tests were expressed as mean ± standard deviation (SD). Means between multiple groups were compared using one-way analysis of variance. Comparisons of means between two groups were performed using independent samples t-tests. Statistical analysis was performed using GraphPad Prism software. Statistical significance was defined as *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Isolation and identification of MSC-EVs and UCB-EVs
TEM results showed that MSC-EVs and UCB-EVs were spherical or disc-shaped (Fig. 1A). NTA results indicated that the particle size range of MSC-EVs and UCB-EVs was between 100 and 200 nm (Fig. 1B). WB results demonstrated that both MSC-EVs and UCB-EVs expressed the EVs surface signature proteins CD63 and TSG101 (Fig. 1C).

Characterization of MSC-EVs and UCB-EVs. (A) The appearance of MSC-EVs and UCB-EVs observed using TEM, with a scale of 100 nm. (B) NTA was used to analyze the particle size distribution of MSC-EVs and UCB-EVs. (C) WB was employed to analyze the expression of CD63 and TSG101.
MSC-EVs and UCB-EVs promote the proliferation and migration of HDF-a
Fluorescence tracing of Exos revealed (Fig. 2A) that both MSC-EVs and UCB-EVs could be phagocytosed and internalized by HDF-a cells when co-cultured for 24 h. CCK-8 assay was used to detect the effects of different concentrations of MSC-EVs and UCB-EVs on the proliferation of HDF-a, and the results showed (Fig. 2B) that cell viability increased with the increase of MSC-EVs and UCB-EVs concentrations in a concentration-dependent manner (P < 0.001). In addition, the effects of MSC-EVs and UCB-EVs (100 µg/mL) on HDF-a proliferation over time were examined, revealing increased HDF-a activity in the MSC-EVs and UCB-EVs treatment groups (P < 0.05) (Fig. 2C). Cell scratch assay results showed that the MSC-EVs and UCB-EVs treatment groups significantly promoted HDF-a migration compared with the control group (P < 0.05) (Fig. 2D, E). The cell migration assay further confirmed these results, showing that the number of migrating HDF-a cells significantly increased in the MSC-EVs and UCB-EVs treatment groups compared with the control group (P < 0.001) (Fig. 2F, G).

MSC-EVs and UCB-EVs promote the proliferation and migration of HDF-a. (A) PKH67-labeled MSC-EVs and UCB-EVs (green fluorescence) are taken up by HDF-a (blue fluorescence ), with specific scale of 50 μm and 100 μm. (B, C) Concentration gradient and time gradient detection of MSC-EVs and UCB-EVs by CCK-8 showed that HDF-a proliferation was promoted. (D) Cell scratch assay showed that MSC-EVs and UCB-EVs promoted HDF-a migration, with a scale of 200 μm. (E) Quantitative analysis of the migration area in figure (D), n = 3. (F) Cell migration assay showed that MSC-EVs and UCB-EVs induced an increase in the motility of HDF-a, compared with the scale: 100 μm. (G) Quantitative analysis of the number of migrating cells in figure (F). *P < 0.05, **P < 0.01, ***P < 0.001.
MSC-EVs and UCB-EVs promote skin wound healing in mice
In vivo experiment results showed that the wound area measured on days 3, 7, 11, 15, and 17 after injury was smaller in the MSC-EVs and UCB-EVs treatment groups than in the control group (Fig. 3A). Quantitative analysis of wound areas at different times and treatments showed that MSC-EVs and UCB-EVs significantly promoted wound healing, with significant differences observed on days 7, 11, 15, and 17 (P < 0.05) (Fig. 3B). HE results showed that the skin tissues of the MSC-EVs and UCB-EVs treated groups had longer new epidermis and dermis, better cuticle repair, and thicker epidermis than those of the control group. Additionally, a large number of lymphocyte infiltrates, capillary hyperplasia, and hair follicle formation were seen in the dermis Of the MSC-EVs and UCB-EVs treated groups(Fig. 3C). Quantitative analysis of the re-epithelialization rate and scar width further confirmed that MSC-EVs and UCB-EVs enhanced epidermal regeneration and reduced wound scar formation (P < 0.05) (Fig. 3E). Masson’s staining indicated more undulating collagen fibers in the skin tissue of the MSC-EVs and UCB-EVs treated groups than in the control group (Fig. 3D). qRT-PCR results showed that MSC-EVs and UCB-EVs significantly increased the expression of Type III collagen and TGF-β3 mRNA in HDF-a and mouse skin tissues, while decreasing the expression of Type I collagen and TGF-β1 mRNA (P < 0.05) (Fig. 3F, G).

MSC-EVs and UCB-EVs promote skin wound healing in mice. (A) Gross views of wounds treated with MSC-EVs, UCB-EVs, or PBS on days 0, 3, 7, 11, 15, and 17 post-injury. (B) Wound healing rates in different treatment groups. Each group contains n = 6. (C) HE staining of wound sections treated with MSC-EVs, UCB-EVs, or PBS 17 days after surgery. The black arrow indicates the edge of the scar. Scale: 500 μm. (D) Masson’s trichrome olor staining of wound sections treated with MSC-EVs, UCB-EVs, or PBS. (E) Quantitative analysis of the scar width and degree of re-epithelialization. Each group contains n = 3. (F) qRT-PCR was used to detect the mRNA expression of scarring-related factors and collagen after co-culture of MSC-EVs and UCB-EVs with HDF-a. (G) qRT-PCR was used to detect the mRNA expression of scarring-related factors and collagen in wound tissue of mice on day 17 after treatment. *P < 0.05, **P < 0.01, ***P < 0.001.
Generation of ST maps of skin tissue after wound healing in mice
ST analysis of mouse wound healing tissue identified several main cell types through the detection of marker genes: stromal cells (Sfrp2), keratinocytes (Flg), hair follicle cells (Myl4), sebocytes (Acacb), and melanocytes (Tyrp1). The localization of each cell group on tissue samples was displayed on HE sections. Additionally, the cell populations of different treatment groups were visualized in the two-dimensional space using UMAP maps (Fig. 4A, B, D, E). The cell proportion diagram showed that the proportion of stromal cells and keratinocytes was reduced in the MSC-EVs and UCB-EVs treated groups compared with the control group. Sebocytes and melanocytes were absent or few in the control group, and hair follicle cells were only present in the control group (Fig. 4C). The relative expression of MB marker genes showed that the relative expression of Acta2, Pdgfra, and Engrailed-1 genes was decreased in both the MSC-EVs and UCB-EVs treatment groups compared with the control group (Fig. 4F, G).

ST shows the major cell types in the skin tissue of mice in different treatment groups. (A) Using 10×Genomics Visium ST sequencing, the results were presented on HE-stained sections of skin samples from different treatment groups. (B) UMAP shows the clustering results of samples from different processing groups. (C) Bar chart shows the proportion and count of each cell population in samples from different treatment groups. (D) Dot plot of marker gene expression in each cell group. (E) Violin map of marker gene expression in each cell group. (F) UMAP shows the expression of MFBs marker genes in different treatment groups. (G) The results of MFBs marker genes on HE-stained sections of skin samples of mice in different treatment groups.
Cell heterogeneity and intercellular communication in skin tissue after wound healing in mice
Gene heat maps showed the differentially expressed top 10 genes in keratinocytes of the MSC-EVs and UCB-EVs treated groups compared with the control group, respectively (Fig. 5A). GSVA pathway enrichment analysis showed that the TGF-β signaling pathway was downregulated in the MSC-EVs treatment group, and the Wnt signaling pathway was upregulated in the UCB-EVs treatment group compared with the control group (Fig. 5B). Gene heat maps of stromal cells showed the differentially expressed top 10 genes in the MSC-EVs and UCB-EVs treated groups compared with the control group, respectively (Fig. 5C). GSVA pathway enrichment analysis showed that the Wnt signaling pathway was upregulated in the MSC-EVs and UCB-EVs treatment groups compared with the control group (Fig. 5D). Analysis of keratinocyte-stromal cell interactions revealed a higher number of cell communication ligand receptors and stronger communication intensity in the MSC-EVs and UCB-EVs treated groups than in the control group (Fig. 6A). The results of ECM ligand-receptor interaction showed that the signals significantly altered in the MSC-EVs and UCB-EVs treatment groups included signals related to fibrosis and ECM remodeling, such as Col1a2, Col1a1, Col6a3, Thbs1, Col4a1, Fn1, and Col3a1 (Fig. 6B, C).

Cell heterogeneity in mouse skin tissue. (A) Heat maps of differentially expressed genes of the top 10 keratinocytes between groups. (B) GSVA pathway enrichment analysis of upregulated genes and wound healing and scar formation-related pathways in MSC-EVs and UCB-EVs treated groups. (C) Heat maps of the top 10 differentially expressed genes in stromal cells between groups. (D) GSVA pathway enrichment analysis of upregulated genes and wound healing and scar formation-related pathways in MSC-EVs and UCB-EVs treated groups.

Cell intercellular communicationin mouse skin tissue. (A) Histogram of the number and intensity of cell communication receptor pairs in each group. (B) The keratinocyte/stromal cell ligand-receptor interaction bubble map in the MSC-EVs treatment group compared with the CT group (the vertical coordinate is the receptor gene pair; the color of the bubble ranges from blue to yellow to red, representing the communication probability; and the bubble size represents the communication significance). (C) Bubble map of keratinocyte/stromal cell ligand-receptor interaction in the UCB-EVs group compared with the control group.
Discussion
Recent studies have shown that EVs, as important components of the cell paracrine pathway, have great potential in promoting skin wound repair and regeneration. Adipose-derived mesenchymal stromal cell-derived EVs (ADMSC-EVs) can promote the proliferation and migration of FBs and optimize collagen deposition through the PI3K/Akt signaling pathway, thereby accelerating wound healing23. miRNAs carried by MSC-EVs may be important regulators of the TGF-β/SMAD signaling pathway, which can inhibit the differentiation of MFBs during skin wound healing, reducing scar formation and promoting skin wound healing24. miR-192-5p in ADMSC-EVs can reduce fibrosis in activated FBs, inhibit the expression of pro-fibrotic proteins, and reduce scar formation through the miR-192-5p/IL-17RA/Smad axis22. The process of tissue repair involves the proliferation, differentiation, migration, apoptosis, and disappearance of various repair cells, with FBs being the main effector cells. Their proliferation and migration are crucial for wound contraction, collagen synthesis, and tissue remodeling25. In this study, we evaluated the effects of MSC-EVs and UCB-EVs on the behavior of HDF-a in vitro and found that these extracellular vesicles could be internalized by HDF-a and significantly enhance their proliferation and migration, suggesting that MSC-EVs and UCB-EVs stimulate wound healing through fibroblast activation.
We established a skin wound model with 6-8-week-old BALB/c mice. MSC-EVs, UCB-EVs, or PBS was injected subcutaneously around the wound from day 3 for 5 consecutive days, and the wound was photographed during the healing process. The results showed that the wound area measured on days 3, 7, 11, 15, and 17 after injury was smaller in the MSC-EVs and UCB-EVs treatment groups than in the control group, and the wound healing process was significantly accelerated. Histological analysis showed that the MSC-EVs and UCB-EVs groups had a greater degree of re-epithelialization, smaller scar width, and more undulating fibers than the control group. Exos from bone mesenchymal stem cells, stimulated by Fe3O4 nanoparticles and a static magnetic field, have been reported to accelerate skin wound healing in rats by upregulating miR-21-5p, narrowing scar width, and enhancing angiogenesis26. Plasma endothelial cell (EC)-derived EVs prevented fibroblast senescence and accelerated skin wound healing in diabetic mice by decreasing YAP phosphorylation and activating the PI3K/Akt/mTOR pathway27. These results indicate that MSC-EVs and UCB-EVs can effectively promote skin wound healing and reduce scar width, with consistent efficacy between the two Exos types.
Scar-free wound tissue is characterized by reticular collagen formation, less crosslinking, less inflammation, and fewer MFBs28. Scar-free wound tissue also has a higher proportion of Type III and Type I collagen, TGF-β3 and TGF-β1, and matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs (TIMPs)29. Type I collagen is mainly found in connective tissue, and its excessive deposition can lead to scarring30. The TGF-β subtype is an important factor involved in ECM deposition and scarring. During wound healing, elevated levels of TGF-β1 promote wound healing primarily by promoting fibroblast activation and collagen synthesis; however, its excessive levels can lead to excessive collagen deposition31, resulting in scarring. TGF-β3 can reduce collagen synthesis and increase the secretion of MMPs, promoting ECM degradation and acting as a negative regulator of the MFBs phenotype32. In this study, MSC-EVs and UCB-EVs increased Type III collagen and TGF-β3 mRNA expression and decreased Type I collagen and TGF-β1 mRNA expression in HDF-a and mouse skin tissues, suggesting that MSC-EVs and UCB-EVs reduce scar formation by regulating collagen deposition and the ratio of TGF-β3 to TGF-β1.
Wound contraction is a sign of wound healing. A special population of FBs, called MFBs, promote tissue contraction33. MFBs increase in number and are crucial for ECM production in fibrotic diseases34. Normal human dermal FBs can be divided into four subgroups: secretion-papillary, secretion-reticular, mesenchymal, and pro-inflammatory35. α-SMA-positive MFBs are abundant in hypertrophic scars; these cells contract, which causes the scar to contract. An ST analysis of wound tissue at different times defined wound healing fibroblast phenotypes as mechanical fibrosis (key fibrosis-related genes such as Col1a1, Acta2, Pdgfra, FAK [Ptk2], and its downstream gene Jun), activator (Fn1 and Thbs1), proliferator (Jak2 upregulated and Fsp1 [S100a4] and Il6 downregulated), and remodeling agents (Ptk2b, Jak1, and Jak3). Known markers related to mechanical fibrosis, such as Engrailed-1, Col1a1, Tgfβ2, and Jun, were highly expressed in mechanofibrotic cells18. After defining keloid FBs by single-cell RNA-seq, the percentage of mesenchymal FBs subpopulations in keloid was significantly increased. To examine the relationship between MFBs and mesenchymal FBs, we analyzed the expression of ACTA2 (α-SMA), a marker gene of MFBs, and found that MFBs were enriched in the mesenchymal FBs subpopulation in keloids36. In this study, ST analysis was performed on the skin tissues of mice in each group after wound healing to elucidate the spatial localization of MFBs marker genes Acta2, Pdgfra, and Engrailed-1, which revealed that MSC-EVs and UCB-EVs could reduce their relative expression levels, thereby reducing scar formation.
Wound healing is a dynamic process in which various types of cells, cytokines, and ECM interact, with keratinocytes and stromal cells being the main cells involved in wound healing and scar formation. For example, activins, members of the TGF family, regulate skin morphogenesis and wound repair in a concentration-dependent manner via keratinocytes and stromal cells, underscoring the importance of epithelial-mesenchymal interactions in vivo37. We employed ST to display differential gene heat maps of keratinocytes and stromal cells (mainly FBs) and performed GSVA pathway enrichment analysis of pathways associated with wound healing and scar formation. The results showed that the TGF-β signaling pathway was downregulated in the MSC-EVs treated group, and the Wnt signaling pathway was upregulated in the UCB-EVs treated group in keratinocytes. In stromal cells, the Wnt signaling pathway was upregulated in both the MSC-EVs and UCB-EVs treated groups.
The TGF-β and Wnt signaling pathways are the main chemical signaling pathways in wound healing. The TGF-β signaling pathway is necessary for wound repair and regeneration, as the loss of TGF-β expression leads to chronic wound non-healing. For example, the TGF-β/SMAD2/SMAD3 signaling pathway can increase epithelial-to-mesenchymal cell transformation and fibroblast activation38. However, continuous activation of the TGF-β/Smad signaling pathway leads to long-term overactivation of FBs and MFBs, and excessive collagen formation, resulting in abnormal scarring39. The Wnt signaling pathway is a key regulator of embryonic development and wound healing. Activated Wnt/β-catenin signals promote not only the migration and differentiation of epidermal keratinocytes but also angiogenesis, follicle regeneration, and epithelial remodeling, directly promoting skin wound healing40. In Sprague-Dawley rats with full-layer skin wounds, activation of Wnt/β-catenin signaling promoted wound closure by regulating epidermal stem cell proliferation, keratinocyte differentiation and migration, and hair follicle regeneration41. Topical application of exogenous Wnt3a to non-healing wounds enhanced endogenous Wnt signaling and promoted better skin wound healing42. In addition, epidermodermal communication is maintained through a typical Wnt signaling pathway, and activation of Wnt/β-catenin in mouse basal keratinocytes increases FBs proliferation and ECM formation43. However, the Wnt /β-catenin signaling pathway is a major target in fibrotic diseases, reducing scarring in animal skin wound models by applying various Wnt and catenin inhibitors locally44.
In skin tissue repair, the interaction between different cell types is crucial for both wound healing and scar formation45. Therefore, to define the cellular communication landscape between cells in the MSC-EVs and UCB-EVs treated groups versus the control group, we performed keratinocyte-stromal cell interaction analysis using the CellChatDB ligand-receptor database46. The signals significantly altered in the MSC-EVs and UCB-EVs treated groups in ECM ligand-receptor interactions included signals related to fibrosis and ECM remodeling, such as Col1a2, Col1a1, Col6a3, Thbs1, Col4a1, Fn1, and Col3a1. Single-cell transcriptome sequencing of normal and keloid scars showed that fibrosis-related signaling pathway, such as TGF-β1, VEGF, and POSTN, were significantly altered in keloids compared with normal scars. Moreover, fibrotic signaling interactions in keloids, such as TGF-β, POSTN, and PDGF, were significantly increased47. Signaling cross-talk between keloid ECs and FBs using NicheNet predicted that keloid ECs regulate keloid FBs by ligands associated with mesenchymal activation, such as COL4A1, SERPINE1, ADAM15, COL1A1, and FN1, compared with normal skin ECs48.
In conclusion, our results suggest that MSC-EVs and UCB-EVs can effectively promote skin wound healing and reduce scar formation in mice possibly via FBs activation at the wound site. In vitro experiments revealed that MSC-EVs and UCB-EVs can enhance the proliferation and migration of HDF-a and increase the proportion of Type III collagen/Type I collagen and TGF-β3/TGF-β1 in FBs and mouse skin tissues. In vivo experiments demonstrated that MSC-EVs and UCB-EVs can significantly improve the degree of re-epithelialization and reduce scar width. ST analysis showed that MSC-EVs and UCB-EVs could alter the functional heterogeneity of major cell types in skin tissue, change the major pathways related to wound healing and scar formation (TGF-β and Wnt signaling pathways), and reduce the relative expression of marker genes in MFBs. Cell-cell interaction analysis determined that MSC-EVs and UCB-EVs altered signaling related to fibrosis and ECM remodeling between keratinocytes and stromal cells. These results suggest that MSC-EVs and UCB-EVs are promising treatments that can promote wound healing and reduce scarring.
Data availability
The sequencing data have been deposited in the NCBI Sequence Read Archive (SRA) database under the accession code PRJNA1163042. https://dataview.ncbi.nlm.nih.gov/object/PRJNA1163042?reviewer=3krhe814t1bga95ue107ngcnkg.
Abbreviations
- EVs:
-
Extracellular vesicles
- miRNAs:
-
microRNAs
- MSC-EVs:
-
Mesenchymal stem cell-derived extracellular vesicles
- UCB-EVs:
-
Human umbilical cord plasma-derived extracellular vesicles
- ST:
-
Spatial transcriptomics
- HDF-a:
-
Human dermal fibroblast-adult cell
- FBs:
-
Fibroblasts
- ECM :
-
Extracellular matrix
- Exos:
-
Exosomes
- EPCs:
-
Endothelial progenitor cells
- EPSC-Exos:
-
Epidermal stem cell-derived Exos
- MFBs:
-
Myofibroblasts
- hAFSCs:
-
Human amniotic fluid stem cells
- TEM:
-
Transmission electron microscopy
- NTA:
-
Anoparticle tracking analysis
- WB:
-
Western blot
- qRT-PCR:
-
Quantitative real-time PCR
- ADMSC-EVs:
-
Adipose-derived mesenchymal stromal cell-derived EVs
- EC:
-
Endothelial cell
- MMPs:
-
Matrix metalloproteinases
- TIMPs:
-
Tissue inhibitors of MMPs
References
Kato, J. et al. Mesenchymal stem cells ameliorate impaired wound healing through enhancing keratinocyte functions in diabetic foot ulcerations on the plantar skin of rats. J. Diabetes Complicat. 28(5), 588–595 (2014).
Plikus, M. V. et al. Regeneration of fat cells from myofibroblasts during wound healing. Science 355(6326), 748–752 (2017).
Sorg, H. et al. Skin wound healing: An update on the current knowledge and concepts. Eur. Surg. Res. 58, 81–94 (2017).
Yao, F. & Eriksson, E. Gene therapy in wound repair and regeneration. Wound Repair. Regen. 8(6), 443–451 (2000).
Rippa, A. L., Kalabusheva, E. P. & Vorotelyak, E. A. Regeneration of dermis: Scarring and cells involved. Cells 8(6), 607 (2019).
Adams, D. C. & Ramsey, M. L. Grafts in dermatologic surgery: Review and update on full- and split-thickness skin grafts, free cartilage grafts, and composite grafts. Dermatol. Surg. 31, 1055–1067 (2005).
Loreti, E. H. et al. Use of laser therapy in the healing process: A literature review. Photomed. Laser Surg. 33(2), 104–116 (2015).
Lichtman, M. K., Otero-vinas, M. & Falanga, V. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Repair. Regen. 24(2), 215–222 (2016).
Eckermann, C. W. et al. Characterization and modulation of fibroblast/endothelial cell co-cultures for the in vitro preformation of three-dimensional tubular networks. Cell Biol. Int. 35(11), 1097–1110 (2011).
Orgill, D. P. & Ogawa, R. Discussion: The embrace device significantly decreases scarring following scar revision surgery in a randomized controlled trial. Plast. Reconstr. Surg. 133(2), 406–407 (2014).
Vlassov, A. V. et al. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 1820(7), 940–948 (2012).
Liao, J. et al. Expression profiling of exosomal miRNAs derived from human esophageal cancer cells by Solexa high-throughput sequencing. Int. J. Mol. Sci. 15(9), 15530–15551 (2014).
Pegtel, D. M. & Gould, S. J. Exosomes. Annu. Rev. Biochem. 88, 487–514 (2019).
Zhang, J. et al. Exosomes derived from human endothelial progenitor cells accelerate cutaneous wound healing by promoting angiogenesis through Erk1/2 signaling. Int. J. Biol. Sci. 12(12), 1472–1487 (2016).
Zhang, B. et al. HucMSC-exosome mediated-Wnt4 signaling is required for cutaneous wound healing. Stem Cells 33(7), 2158–2168 (2015).
Duan, M. et al. Epidermal stem cell-derived exosomes promote skin regeneration by downregulating transforming growth factor-β1 in wound healing. Stem Cell Res. Ther. 11(1), 452 (2020).
Zhang, Y. et al. Human amniotic fluid stem cell-derived exosomes as a novel cell-free therapy for cutaneous regeneration. Front. Cell Dev. Biol. 9, 685873 (2021).
Foster, D. S. et al. Integrated spatial multiomics reveals fibroblast fate during tissue repair. Proc. Natl. Acad. Sci. U.S.A. 118(41), e2110025118 (2021).
Goodarzi, P. et al. Mesenchymal stem cells-derived exosomes for wound regeneration. Adv. Exp. Med. Biol. 1119, 119–131 (2018).
Hu, Y. et al. Exosomes from human umbilical cord blood accelerate cutaneous wound healing through mir-21-3p-mediated promotion of angiogenesis and fibroblast function. Theranostics 8(1), 169–184 (2018).
van Balkom, B. W. et al. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 121(19), 3997–S15 (2013).
Li, Y. et al. Exosomes derived from human adipose mesenchymal stem cells attenuate hypertrophic scar fibrosis by miR-192-5p/IL-17RA/Smad axis. Stem Cell Res. Ther. 12(1), 221 (2021).
Zhang, W. et al. Cell-free therapy based on adipose tissue stem cell-derived exosomes promotes wound healing via the PI3K/Akt signaling pathway. Exp. Cell Res. 370(2), 333–342 (2018).
Fang, S. et al. Umbilical cord-derived mesenchymal stem cell-derived exosomal micrornas suppress myofibroblast differentiation by inhibiting the TGF-β/Smad2 pathway during wound healing. Stem Cells Transl. Med. 5(10), 1425–1439 (2016).
Driskell, R. R. et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 504(7479), 277–281 (2013).
Wu, D. et al. Exosomes Derived from Bone mesenchymal stem cells with the stimulation of Fe3O4 nanoparticles and static magnetic field enhance Wound Healing through upregulated miR-21-5p. Int. J. Nanomed. 15, 7979–7993 (2020).
Wei, F. et al. Plasma endothelial cells-derived extracellular vesicles promote wound healing in diabetes through YAP and the PI3K/Akt/mTOR pathway. Aging (Albany NY) 12(12), 12002–12018 (2020).
Yates, C. C., Hebda, P. & Wells, A. Skin wound healing and scarring: Fetal wounds and regenerative restitution. Birth Defects Res. C Embryo Today 96(4), 325–333 (2012).
Zheng, Z. et al. Fibromodulin is essential for fetal-type scarless cutaneous wound healing. Am. J. Pathol. 186(11), 2824–2832 (2016).
Wang, L. et al. Author correction: Exosomes secreted by human adipose mesenchymal stem cells promote scarless cutaneous repair by regulating extracellular matrix remodelling. Sci. Rep. 11(1), 3245 (2021).
Huh, M. I. et al. Distribution of TGF-beta isoforms and signaling intermediates in corneal fibrotic wound repair. J. Cell. Biochem. 108(2), 476–488 (2009).
Occleston, N. L. et al. Prevention and reduction of scarring in the skin by transforming growth factor beta 3 (TGFbeta3): From laboratory discovery to clinical pharmaceutical. J. Biomater. Sci. Polym. Ed. 19(8), 1047–1063 (2008).
Desmoulière, A., Guyot, C. & Gabbiani, G. The stroma reaction myofibroblast: A key player in the control of tumor cell behavior. Int. J. Dev. Biol. 48(5–6), 509–517 (2004).
Schuster, R. et al. The role of myofibroblasts in physiological and pathological tissue repair. Cold Spring Harb Perspect. Biol. 15(1), a041231 (2023).
Solé-boldo, L. et al. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 3(1), 188 (2020).
Andrews, J. P. et al. eloids: The paradigm of skin fibrosis—Pathomechanisms and treatment. Matrix Biol. 51, 37–46 (2016).
Bamberger, C. et al. Activin controls skin morphogenesis and wound repair predominantly via stromal cells and in a concentration-dependent manner via keratinocytes. Am. J. Pathol. 167(3), 733–747 (2005).
Wang, J. et al. Deep dermal fibroblasts contribute to hypertrophic scarring. Lab. Invest. 88(12), 1278–1290 (2008).
Zhang, T. et al. Current potential therapeutic strategies targeting the TGF-β/Smad signaling pathway to attenuate keloid and hypertrophic scar formation [J]. Biomed. Pharmacother. 129, 110287 (2020).
Birdsey, G. M. et al. The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/β-catenin signaling. Dev. Cell 32(1), 82–96 (2015).
Shi, Y. et al. Wnt and notch signaling pathway involved in wound healing by targeting c-Myc and Hes1 separately. Stem Cell Res. Ther. 6(1), 120 (2015).
Whyte, J. L. et al. Augmenting endogenous wnt signaling improves skin wound healing. PLoS ONE 8, 10e76883 (2013).
Ansell, D. M. et al. Exploring the hair growth-wound healing connection: anagen phase promotes wound re-epithelialization. J. Invest. Dermatol. 131(2), 518–528 (2011).
Leavitt, T. et al. Scarless wound healing: Finding the right cells and signals. Cell. Tissue Res. 365(3), 483–493 (2016).
Werner, S., Krieg, T. & Smola, H. Keratinocyte-fibroblast interactions in wound healing. J. Invest. Dermatol. 127(5), 998–1008 (2007).
Jin, S. et al. Inference and analysis of cell–cell communication using CellChat. Nat. Commun. 12(1), 1088 (2021).
Deng, C. C. et al. Single-cell RNA-seq reveals fibroblast heterogeneity and increased mesenchymal fibroblasts in human fibrotic skin diseases [J]. Nat. Commun. 12 (1), 3709 (2021).
Shim, J. et al. Integrated Analysis of Single-Cell and spatial transcriptomics in keloids: Highlights on fibrovascular interactions in keloid pathogenesis. J. Invest. Dermatol. 142(8), 2128–2139e11 (2022).
Acknowledgements
We thank the Lactation Biology Team of the College of Veterinary Medicine of Henan Agricultural University for their guidance and assistance. This work was supported by a grant from HenanYinfeng Biological Engineering Technology Co., LTD.
Author information
Authors and Affiliations
Contributions
Ruonan Li wrote the manuscript; Ruonan Li, Kai Zhong, Xiaolong Wang. and Xuemei Zhang designed and supervised research; Ruonan Li and Hantian Wang performed the experiments and analyzed data; Ruonan Li, Yanbin Yang, and Heping Li managed data; all authors proofread the manuscript and approved the final version of this paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This project (title: Research on MSC-EVs and UCB-EVs Promoting Skin Wound Healing and Reducing Scar Formation) were approved by the Animal Research Committee of Henan Agricultural University and all animals were maintained in a specific pathogen-free animal facility according to the related ethical regulations instilled at Henan Agricultural University and the guide for the care and use of laboratory animals. (Name of the institutional approval committee or unit: Science Ethics Committee of Henan Agricultural University; Approval Number: HNND2024092901; Approval Date: 4 March 2023). The mesenchymal stem cells supernatant and cord blood used in this study were provided by Henan Yinfeng Bioengineering Technology Co., LTD. The project (Title: Henan Cell and Tissue Human Genetic Resources Bank Preservation / Biobank) has been approved by the Medical Ethics Committee of Nanyang First People’s Hospital. (Name of the institutional approval committee or unit: Medical Ethics Committee of Nanyang First People’s Hospital; Approval Number: [2023-yx111z] 0621-01; Approval Date: June 19, 2023).
Consent for publication
Written informed consent for publication was obtained from all participants.
Artificial intelligence
The authors declare that they have not use AI-generated work in this manuscript
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, R., Wang, H., Wang, X. et al. MSC-EVs and UCB-EVs promote skin wound healing and spatial transcriptome analysis. Sci Rep 15, 4006 (2025). https://doi.org/10.1038/s41598-025-87592-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-87592-6
