
Abstract
Heart failure (HF) is a complex and prevalent condition, particularly in the elderly, presenting symptoms like chest tightness, shortness of breath, and dyspnea. The study aimed to improve the classification of HF subtypes and identify potential drug targets by exploring the role of Immunogenic Cell Death (ICD), a process known for its role in tumor immunity but underexplored in HF research. Additionally, the study sought to apply deep learning models to enhance HF classification and identify diagnosis-related genes. Various deep learning encoder models were employed to evaluate their effectiveness in clustering HF based on ICD-related genes. Identified HF subtypes were further refined using differentially expressed genes, allowing for the assessment of immune infiltration and functional enrichment. Advanced machine learning techniques were used to identify diagnosis-related genes, and these genes were used to construct nomogram models. The study also explored gene interactions with miRNA and transcription factors. Distinct HF subtypes were identified through clustering based on ICD-related genes. Differentially expressed genes revealed significant variations in immune infiltration and functional enrichment across these subtypes. The diagnostic model showed excellent performance, with an AUC exceeding 0.99 in both internal and external test sets. Diagnosis-related genes were also identified, serving as the foundation for nomogram models and further exploration of their regulatory interactions. This study provides a novel insight into HF by combining the exploration of ICD, the application of deep learning models, and the identification of diagnosis-related genes. These findings contribute to a deeper understanding of HF subtypes and highlight potential therapeutic targets for improving HF classification and treatment.
Similar content being viewed by others

Introduction
Heart failure (HF) is a clinical syndrome characterized by impaired cardiac ejection or filling functions resulting from the interplay of multiple pathological factors. Its primary clinical manifestations include dyspnea, angina, and dizziness1,2. As the structural and functional deterioration of the heart progresses, the development of HF is often accompanied by morphological changes in cardiomyocytes and dysregulation of energy metabolism. These complications, particularly in the elderly, exacerbate late-stage HF and increase the challenges in its diagnosis and treatment3,4. Consequently, there is an urgent need to identify biomarkers associated with HF for improved diagnosis, risk assessment, and therapeutic targeting, aiming to enable early intervention and personalized treatment5.
Inflammation and immune dysregulation have emerged as key factors in the pathogenesis and progression of heart failure (HF). Chronic low-grade inflammation, immune cell infiltration, and dysregulated cytokine signaling characterize the microenvironment of HF, contributing to adverse remodeling and cardiac dysfunction6,7. Among emerging immune processes, immunogenic cell death (ICD) has garnered significant attention. Unlike apoptosis or necrotic cell death, ICD activates adaptive immune responses by releasing damage-associated molecular patterns (DAMPs) and promoting antigen presentation by dendritic cells, ultimately stimulating cytotoxic T-cell activation8,9. However, the relationship between ICD-related genes (ICDRGs) and cardiovascular diseases,including HF, remains poorly understood. The inflammatory and immunological features of HF suggest potential overlap with immune responses mediated by ICD. Investigating the role of ICDRGs in HF could provide insights into novel immune-mediated mechanisms and their diagnostic and therapeutic implications. Furthermore, as ICD is known to shape the immune microenvironment, studying ICDRGs may uncover potential biomarkers for HF subtype classification and prognosis.
This study aims to comprehensively investigate the relationship between ICDRGs and HF, focusing on their potential diagnostic and subtype classification applications. Gene expression data for HF and normal control groups were retrieved from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). Differentially expressed genes were identified and intersected with ICDRGs, followed by bioinformatics analyses of the intersecting genes. HF subtypes were identified using three clustering methods, with subsequent exploration of pathway enrichment and immune system differences among subtypes. Further subtype stratification was performed using differentially expressed genes, coupled with immune cell infiltration and functional enrichment analyses. In addition, two machine learning algorithms were employed to identify key genes associated with HF diagnosis, leading to the construction of a diagnostic nomogram model. The correlation between these diagnostic genes and immune cell types was also investigated. Finally, miRNA-mRNA interaction networks and transcription factor regulatory networks related to diagnostic genes were constructed. This study seeks to elucidate the potential roles and mechanisms of ICDRGs in HF, providing novel insights and evidence for personalized therapy and risk assessment in HF.
Method
Clustering algorithm
Autoencoders
Autoencoders (AE) is a deep neural network that consists of an encoder and a decoder. Both the encoder and decoder are composed of multilayer feedforward neural networks. They are connected by the bottleneck layer. The encoder and decoder are represented by Formula (2) and Formula (3), respectively.
where \(z\) is the output of the encoder, which can be regarded as a reduced-dimension representation of data. x is the output of the decoder. \({f}_{\text{encoder}}\) and \({f}_{\text{decoder}}\) are multilayer neural networks. In this paper, all encoder-based models are implemented using pytorch. Among them, the loss function used by AE is MSE loss functions. For all encoder parts, the number of network layers was set to [10, 5, cluster_num]. Cluster_num represents the number of clusters. For all decoder parts, the network layer number was set to [10, 5, cluster_num]. For all models based on the self-encoder, epoch was set to 100 during training.
Denoising autoencoders
Different from AE, denoising autoencoders (DAE) constructs partially damaged data by adding noise to the input data, and then restores it to the original input data by encoding and decoding. The newly generated \(\tilde{x}\) can be expressed by the following formula.
where \({q}_{D}\) represents random mapping and obeys the unit normal distribution \(\mathcal{N}(0,1)\). Its encoder and decoder can be represented by Formula (4) and Formula (5), respectively.
K-means clustering algorithm
K-means clustering is a classical clustering algorithm, and its implementation steps are as follows: Firstly, k clustering centers are randomly selected. Then the distance from each sample point is calculated, and the cluster center will divide it into the nearest cluster, forming k clusters. Next, the centroid (mean) of each cluster is recalculated. Repeat the above process until the position of the center of mass no longer changes or reaches the set number of iterations. In this paper, the algorithm was implemented with default parameters based on the scikit-learn package of Python.
Algorithm evaluation index
In this paper, three evaluation indexes of clustering performance were introduced, including the sample contour coefficient (the value was between −1 and 1, the closer to 1, the better the clustering effect), the Calinski-Harabasz Index (the value was greater than 0, the better the clustering effect), and the Davies-Bouldin Index (the value was greater than 0, the closer to 0, the better the clustering effect). All of them were implemented by Python’s scikit-learn package.
Algorithm input and parameter settings
First, the expression profiles of the specified genes in the HF group were used as input data. AE and DAE models were constructed to extract nonlinear features, with the hidden layer dimensions set to2,6,10. The noise factor for the DAE was set to 0.3, and the training parameters were configured as epochs = 100 and batch_size = 4. The output features of the models were clustered using KMeans, and performance was evaluated. For other algorithms, the default parameters of the Python "scikit-learn" package were used. Additionally, KMeans and VAE were directly applied for clustering analysis. High-resolution scatter plots were generated using t-SNE for dimensionality reduction to visualize the results.
Data acquisition
All the data in this paper came from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). Specifically,we used GSE141910 data set (126 diseased samples and 240 control samples) as the training set, and the GSE116250 data set (50 diseased samples and 14 control samples) as the test set. The GSE141910 data set is derived from left ventricular free wall tissue harvested during cardiac surgery from HF subjects undergoing transplantation and from unused donor hearts with apparently normal function. Cold myocardial paralysis was perfused before cardiac resection to block contraction and prevent ischemic damage, and tissue specimens were frozen in liquid nitrogen. The GSE141910 data set comes from 64 samples of human left ventricular tissue. We standardized the data using the normalizeBetweenArrays function in the limma package, and then removed genes with zero expression levels.
The expression of ICDRGs before and after renal ischemia–reperfusion
The differential expression analysis of the GSE141910 data set was carried out using the “limma” package, and 8885 differentially expressed genes were obtained. We set Adj.P.Val < 0.05 as the threshold for screening differentially expressed genes. Then, this paper collected 20 ICDRGs from the previous work and intersected them with differentially expressed genes to obtain intersection genes (DEICDRGs). The expression of ICDRGs in the diseased group and the control group in the GSE141910 data set was displayed as a box graph. In order to evaluate the correlation between ICDRGs, the Pearson correlation coefficient of DEICDRGs in the sample is calculated and visualized by “corrplot” in R software.
Enrichment analysis of different clusters
In this paper, the enrichment analysis of gene ontology (GO) and genome encyclopedia (KEGG) was realized by using the R package "clusterProfiler."And the analysis of gene set variation (GSVA) was realized by using R-packet "GSVA." Among them, c2.cp.kegg.v7.4.symbols.gmt was downloaded from the MSigDB database and used to study the changes in biological signal pathways10,11,12. The R package “ggplot2” was used to visualize the enrichment results. A p-value less than 0.05 is considered statistically significant.
Immunoassay
The ssGSEA algorithm was used to estimate the infiltration abundance of immune cells and the score of immune function in the sick group and the control group. Then, we also collected the genes related to immune inspection sites and explored the difference in expression levels of genes related to immune inspection sites between the two groups by box chart.
Construction and verification of HF-related diagnosis model
For the purpose of screening the diagnosis-related genes of HF, we adopt a small absolute contraction and selection operator (LASSO) and support vector machine recursive feature elimination (SVM-RFE). The R-packet “glmnet” was used to implement the LASSO algorithm in the case of ten times cross-validation. SVM-RFE was also implemented by R script under the condition of ten times cross-validation. Finally, we draw ROC curves of diagnostic genes and diagnostic models by using the R-package "pROC."
Construction of nomogram
The expression matrix of diagnostic-related genes was used as the input for the nomogram. The study constructed the nomogram model based on diagnosis-related genes using the R package “rms”. Then the validity of the nomogram model was evaluated by the calibration curve. The clinical practicability of the nomogram model was evaluated by a decision curve. Finally, the high-risk probability stratification was predicted by the clinical impact curve when the population size was 1000.
Construction of interaction network
We conducted online analysis of hub genes using the NetworkAnalyst database (https://www.networkanalyst.ca/NetworkAnalyst/) to construct a transcription factor (TF) hub gene network and a miRNA hub gene interaction network. In this research, miRNA and TF interacting with diagnosis-related genes were queried by using the miRTarBase database (http://mirtarbase.cuhk.edu.cn/php/index.php) and TargetScan database (http://www.targetscan.org/vert_72/) respectively. Then, an interaction network between diagnostic-related genes and miRNA was constructed, the same between diagnostic-related genes and TF.
Experimental validation of diagnosis related related genes
In this paper, the mRNA expression of diagnosis related genes was detected by qRT-PCR. AC16 cell line (sourced from the National Infrastructure of Cell Line Resource in Beijing, China) was used for modeling analysis of heart failure and controls. The mRNA expression level was calculated with the 2-ΔΔCt. All data were expressed by means ± SD, and the statistical differences between groups were tested by T-Test, and p < 0.05 indicated a significant difference. The primer sequences are shown in Table 1.
Results
ICD-related gene expression landscape
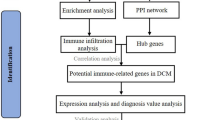
The overall flow chart of this paper is given in Fig. 1. Firstly, the transcriptomic data of the GSE141910 data set were analyzed for differential expression, and 8885 DEGs were obtained with adj.P.Val < 0.05 as the standard. This study collected 20 ICD-related genes from previous literature13. To identify ICD-related genes that were differentially expressed between the HF group and the control group, an intersection was taken between the DEGs and the 20 ICD-related genes, resulting in 14 overlapping genes (Fig. 2A-D). Figure 2E shows the correlation thermogram among 14 DEICDRGs. It was obvious that most genes had a significant correlation. The details of DEGs, ICDRGs, and DEICDRGs can be found in Supplementary Materials (Intersection_case.xlsx).

The technical roadmap of the article.

The expression landscape of DEICDRGs. A. The intersection Wayne diagram of DEGs and ICDRGs (p = 3.409506e-52). B. The expression heat map of DEICDRGs obtained by differential expression analysis. C. The box diagram of the differential expression of DEICDRGs in HF and its control group. D. The chromosome map of DEICDRGs. E. The correlation analysis heat map of DEICDRGs.
Identification of subtypes of HF based on DEICDRGs
The expression levels of 14 DEICDRGs in diseased samples were extracted in the study. We set the number of clusters to 2–5, respectively. Figure 3 displays the tsne dimensionality reduction graphs of AE, DAE, and K-means under different cluster numbers. Each point in the graph represented a sample. In order to evaluate the clustering performance of the three algorithms under different cluster numbers, this paper used three index columns: sample contour coefficient, Calinski-Harabasz Index, and Davies-Bouldin Index. We can observe the histograms of the three algorithms in four clustering situations in Figs. 4A-C. From the figure, we found that DAE had the best comprehensive performance of the three indicators when the number of clusters was 2. Therefore, this paper interpret it as the result of subtype identification in the future. Figures 4D and 4E are box graphs indicating the differences in infiltration abundance and immune function of immune cells between the two types. We also obtained the difference in gene expression related to immune inspection sites between the two subtypes (Fig. 4F). According to Fig. 4F, there were significant differences in the infiltration abundance of most immune cells, immune function, and expression of genes related to immune inspection sites between the two typing samples, which confirmed the typing ability of the DAE algorithm. In addition, this paper also analyzed the GSVA results for two types (Fig. 4G). We analyze the biological significance of these pathways in detail in the discussion section.

Identification process of HF subtypes. A, B, and C are the tsne dimensionality reduction scatter plot obtained by AE, DAE, and K-means algorithms when the number of clusters is set to two categories, respectively. D, E, and F are the tsne dimension reduction scatter plot obtained by three algorithms when the number of clusters is set to three categories, respectively. G, H, and I are the tsne dimension reduction scatter plot obtained by three algorithms when the number of clusters is set to four categories, respectively. J, K, and L are the tsne dimension reduction scatter plot obtained by three algorithms when the number of clusters is set to five categories, respectively.

The performance of algorithm and the analysis of subtypes in the immune microenvironment and functional biological characteristics. A, B, and C are the histogram of the sample contour coefficient, Calinski-Harabasz Index, and Davies-Bouldin Index of three algorithms under four clustering numbers, respectively. D and E are the difference box diagram of infiltration abundance and immune function of immune cells among different types, respectively. F. The box diagram of the difference in immune examination sites between different subtypes. G. The GSVA analysis between subtypes.
The expression levels of 14 DEICDRGs in diseased samples were extracted in the study. We set the number of clusters to range from 2 to 5. Figure 3 displays the t-distributed stochastic neighbor embedding (t-SNE) dimensionality reduction graphs of AE, DAE, and K-means clustering under different cluster numbers. Each point in the figure represented a sample. This paper used three index columns to evaluate the clustering performance of the three algorithms under other cluster numbers: sample silhouette coefficient, Calinski-Harabasz Index, and Davies-Bouldin Index. We can observe the histograms of the three algorithms in four clustering situations in Figs. 4A-C. From the figure, we found that DAE had the best overall performance of the three indicators when the number of clusters was 2. Therefore, this paper interpret it as the outcome of subtype identification in the future. Figures 4D and 4E are box plots indicating the differences in infiltration abundance and immune function of immune cells between the two types. We also obtained the difference in gene expression related to immune inspection sites between the two subtypes (Fig. 4F). According to Fig. 4F, there were significant differences in the infiltration abundance of most immune cells, immune function, and expression of genes related to immune inspection sites between the two subtypes, which confirmed the classification ability of the DAE algorithm. This article introduces non-negative matrix factorization (NMF) and consensus clustering method (CC) to confirm the algorithm’s performance further. Three clustering performance indicators were calculated when the number of clusters was 2. The dimensionality reduction results of these two algorithms and the performance comparison results with the research algorithms included in this article are shown in Figure S1 and Figure S2 in the supplementary material (Supplementary material.docx file), respectively.
In addition, this paper also analyzed the GSVA results for two types (Fig. 4G). Through the GSVA analysis of the two subtypes, we found that there were significant differences in several biological processes between the two subtypes (Fig. 4G). Almost all of these pathways are associated with heart failure—for example, melanoma, apoptosis, and the JAK-STAT signal pathway. Early scholars reported a case where metastatic malignant melanoma could result in rapid occlusion of the right ventricle, thus leading to congestive HF14. Melanoma often involves the heart, resulting in cardiac issues such as HF and myocardial infarction15,16. HF is also intricately linked to the complex pathophysiology of apoptosis17,18. Researchers have confirmed that isoproterenol (ISO) can induce apoptosis, improve heart function, and relieve and treat ISO-induced HF models and cellular HF in rats19. The JAK-STAT signaling pathway has been proven to play an essential role in the pathophysiology of HF20,21.
Verification of subtypes of HF based on DEGs cluster
To further validate the rationality of the above subtype identification, we conducted a differential expression analysis of the two subtypes and identified 43 DEGs with p < 0.05. Using these DEGs, this paper utilized three algorithms to generate t-SNE dimensionality reduction graphs under four kinds of cluster numbers (Fig. 5A-I). We presented the evaluation results of three indicators in Figs. 6A-C. It can be observed that the clustering effect was significantly improved compared with Fig. 5A-l. This confirmed the rationality of using DAE to divide patients into two subtypes. Similarly, we provided the identification results of the immune microenvironment and functional biological characteristics of different gene clusters in Figs. 6D-G. There were noticeable disparities between the two subtypes in the infiltration abundance of immune cells, immune function, the expression of genes related to immune checkpoint sites, and the pathways derived from GSVA analysis.

Re-identification of HF subtypes based on DEGs. A, B and C are the tsne dimensionality reduction scatter plot obtained by AE, DAE, and K-means algorithms when the number of clusters is set to two categories, respectively. D, E, and F are the tsne dimension reduction scatter plot obtained by three algorithms when the number of clusters is set to three categories. G, H, and I are the tsne dimension reduction scatter plot obtained by three algorithms when the number of clusters is set to four categories, respectively. J, K, and L are tsne dimension reduction scatter plot obtained by three algorithms when the number of clusters is set to five categories, respectively.

The performance of algorithm and the analysis of subtypes based on DEGs identification in the immune microenvironment and biological function characteristics. A, B, and C are the histogram of the sample contour coefficient, Calinski-Harabasz Index, and Davies-Bouldin Index of three algorithms under four clustering numbers, respectively. D and E are the difference box diagram of infiltration abundance and immune function of immune cells among different types, respectively. F. The box diagram of the difference in immune examination sites between different subtypes. G. The GSVA analysis between subtypes.
Construction and verification of lasso model and SVM model
This paper extracted diagnosis-related genes from 14 DEICDRGs using the LASSO and SVM-RFE algorithms to identify genes relevant to HF diagnosis. Figures 7A-B display the curves depicting the relationship and cross-validation results of the L1 norm and coefficients obtained by applying the LASSO algorithm. Figure 7C is the result of feature selection using SVM regression. When the number of DEICDRGs was 12, the AUC of tenfold cross-validation was 0.978. Figure 7D is the Venn diagram of the gene intersection screened by LASSO and SVM. We identified 11 genes in the intersection. In the supplementary material (intersection_of_related_diagnosis_genes.xlsx), we provide detailed information on diagnosis-related genes selected by the LASSO algorithm, the SVM-RFE algorithm, and their intersection genes. Figures 7E-F are ROC curves of the diagnostic model constructed using a set of 12 genes in both the training and test sets. Among them, the AUC in the training set reached 0.995 (CI: 0.986–0.999). The AUC in the test set reached 0.95 (CI: 0.841–1), indicating a high level of diagnostic accuracy. In addition, we also evaluated the AUC for each diagnosis-related gene.

The diagnosis model and ROC analysis of the model based on LASSO and SVM. A. The relationship curve between l1 norm and coefficient in the Lasso regression of DEICDRGs. B. The cross-validation result of Lasso regression. C. The result of using SVM regression to filter features. D. The Wayne diagram of the intersection of characteristic genes screened by Lasso and SVM. E and F are ROC analysis of the diagnosis model in the training set and test set, respectively.
Most of the diagnosis-related genes have been shown to play key roles in the development of HF. ATG5 is involved in the formation of autophagic vesicles, which may play an important role in the process of apoptosis. Autophagy is associated with HF, and autophagy activity has been detected in both patients with HF and animal models. The balance between myocardial apoptosis and autophagy in chronic HF can also be treated with drugs22,23. The role of apoptosis in HF has also been determined17,18,19. CASP1 encodes a protein that is a member of the cysteine-aspartic acid protease (caspase) family. Caspases are involved in the signaling pathways of apoptosis, necrosis, and inflammation. IL1R1 is related to immune and inflammatory reactions induced by many cytokines. For a long time, we have been concerned that HF is related to systemic inflammation. Essentially, the progress of HF is attributed to the continuous signal transduction of pro-inflammatory cytokines, and the early stage of HF also shows an inflammatory state 34524,25,26. The protein encoded by IL-10 gene is a cytokine that plays a pleiotropic role in immune regulation and inflammation and participates in the regulation of JAK-STAT signaling pathway. TNF encodes a multifunctional pro-inflammatory cytokine belonging to TNF superfamily that is involved in regulating apoptosis. The relationship between HF and TNF was recognized as early as 199027. The mortality of patients with HF increases with the increase in TNF-α level28. TNF family members may represent a new target for HF treatment29.
Figures 8A-K show the ROC curves of the following genes (ATG5 (AUC:0.723,CI:0.67–0.775), CASP1 (AUC:0.883,CI:0.848–0.917), CD8A (AUC:0.703,CI:0.644–0.755), ENTPD1 (AUC:0.663,CI:0.610–0.713), IL1R1 (AUC:0.748,CI:0.697–0.798), IL10 (AUC:0.922,CI:0.893–0.949), IL17RA (AUC:0.842,CI:0.800–0.880), MYD88 (AUC:0.690,CI:0.635–0.742), NT5E (AUC:0.913,CI:0.829–0.942), PRF1 (AUC:0.869,CI:0.829–0.903), and TNF (AUC:0.677, CI:0.619–0.729)) in the training set. Figures 9A-K display the ROC curves of the following genes (ATG5 (AUC:0.723,CI:0.670–0.775), ASP1 (AUC:0.883,CI:0.848–0.917), D8A (AUC:0.703,CI:−0.644–0.755), NTPD1 (AUC:0.663,CI:0.610–0.713), L1R1 (AUC:0.748,CI:0.697–0.798), L10 (AUC:0.922,CI:0.893–0.949), L17RA (AUC:0.842,CI:0.800–0.880), YD88 (AUC:0.690,CI:0.635–0.742), T5E (AUC:0.913,CI:0.879–0.942), RF1 (AUC:0.869,CI:0.829–0.903), and TNF (AUC:0.677,CI:0.619–0.729)) in the test set. All the genes had diagnostic significance for HF.

Diagnostic performance verification of diagnostic genes and diagnostic models in the training set. A-K are ROC curves of ATG5, CASP1, CD8A, ENTPD1, IL1R1, IL10, IL17RA, MYD88, NT5e, PRF1 and TNF in the training set, respectively.

Diagnostic performance verification of diagnostic genes and diagnostic models in the test set. A-K are ROC curves of ATG5, CASP1, CD8A, ENTPD1, IL1R1, IL10, IL17RA, MYD88, NT5e, PRF1, and TNF in the test set, respectively.
Construction of nomogram model
Figure 10 illustrates the development and evaluation of a nomogram model based on diagnostically relevant genes. The input for the nomogram consists of 11 selected genes: ATG5, CASP1, CD8A, ENTPD1, IL1R1, IL10, IL17RA, MYD88, NT5E, PRF1, and TNF. Each gene contributes a specific number of points to the total score based on its expression level (Fig. 10A). The “Points” scale at the top of the nomogram represents the contribution of each gene to the model, where higher scores indicate a stronger association with the predicted risk. The “Total Points” represent the sum of all gene-specific points for an individual patient, which is mapped to a corresponding "Risk of Disease" to estimate the likelihood of the condition. To evaluate the diagnostic accuracy of the nomogram, a calibration curve was generated (Fig. 10B). The curve demonstrates that the predicted probabilities closely align with the actual probabilities, indicating robust model calibration. DCA (Fig. 10C) shows that the nomogram model provides greater clinical utility compared to individual diagnostically relevant genes, particularly across a wide range of threshold probabilities. This result highlights the nomogram’s superior ability to inform clinical decisions. Additionally, the clinical impact curve (Fig. 10D) indicates the number of individuals classified as high-risk based on varying thresholds, as well as the actual number of high-risk individuals who experience the event. This curve further validates the practical application and reliability of the nomogram model for identifying individuals at risk. Finally, we show a scatter plot of correlations between diagnosable genes and immune cell infiltration patterns in Fig. 11A-V.

Construction of column diagram model. A is a nomogram model constructed on selected diagnostically-relevant genes (ATG5, CASP1, CD8A, ENTPD1, IL1R1, IL10, IL17RA, MYD88, NT5E, PRF1, and TNF ). B is the calibration curve used to evaluate the diagnostic capability of the nomogram model. C is based on DCA, and the Nomogram model had higher clinical utility than a single diagnostically-relevant gene. D is the clinical impact curve.

Correlation analysis between diagnosis-related genes and immune cells. A-V are scatter plot of the correlation between diagnostic genes and immune cells/immune function, respectively.
Correlation analysis of immune infiltration and construction of the regulatory network
For the miRNA-mRNA interaction network (Fig. 12A), we confirmed that some miRNAs are related to the pathogenesis of HF by consulting the literature. MiR-423-5p was initially identified as a circulating biomarker of heart disease. Tijsen et al. proved that the circulating level of miR-423-5p in patients with clinical HF increased30. Deng et al. also studied and determined that miR-423-5p is a potential target for the diagnosis and treatment of HF31. The protein coding gene regulated by miR-107 and the gene regulated by miR-139-5p were identified as genes that play a role in HF32. For the miRNA-TF interaction network (Fig. 12B), we know that CREB3 can promote the expression of inflammatory genes. RELA, also known as NF-kappa-B, is a pleiotropic transcription factor, exists in almost all cell types and is the endpoint of a series of signal transduction events, which are triggered by a large number of stimuli related to many biological processes, such as inflammation, immunity, differentiation, cell growth, tumorigenesis, and apoptosis. ATF1 can regulate the expression of downstream target genes to affect cell physiological processes, which is related to soft tissue melanoma, and the relationship between melanoma and HF has been confirmed in previous literature14,15,16.

Exploration of the interaction between diagnosis-related genes and miRNA and TF. A. The miRNA-mRNA interaction network of diagnosis-related genes. B. The mRNA-TF interaction network of diagnosis-related genes.
Results qRT-PCR experimental verification
As shown in Fig. 13, the expression levels of diagnosis-related genes MYD8, TNF, ATG5, CD8A, ENTPD1, IL17RA, NT5E, IL1R1, PRF1, IL-10 and CASP1 in the HF-cell model and control cell model. Among them, the p-value of MYD8, ATG5, ENTPD1, and IL1R1 expression in both groups was less than 0.05. MCAD, CD8A, IL17RA, NT5E, and CASP1 had p-value less than 0.01 in both groups. PRF1 and IL-10 had p-value less than 0.001 in both groups. The expression trends of these genes through qRT-PCR experiments were consistent with the results of differential expression analysis.

A-K are the mRNA expression levesl of MYD8, TNF, ATG5, CD8A, ENTPD1, IL17RA, NT5E, IL1R1, PRF1, IL-10 and CASP1 by qRT-PCR. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Heart failure is a deterioration in heart function resulting from various heart diseases, and patients will show symptoms such as shortness of breath, fatigue, and palpitation. Complex HF often occurs in the elderly and is challenging to diagnose. ICD can elicit various immune reactions, but the function of ICDRGs in HF is still unclear. Therefore, this paper uses three clustering algorithms to identify the subtypes of HF based on ICDRGs. The DAE model is divided into two clusters by three clustering indexes for the optimal result. Significant differences exist between the two subtypes in the infiltration levels of immune cells, immune function, and the expression of genes related to immune surveillance sites.
We also cluster again according to DEGs between subtypes to verify the reliability of the above clustering results. We found that there were significant differences in the immune microenvironment and functional enrichment among different subtypes of samples obtained by re-clustering. Furthermore, we used the LASSO algorithm and the SVM algorithm to select genes related to the diagnosis of HF (ATG5, CASP1, CD8A, ENTPD1, IL1R1, IL10, IL17RA, MYD88, NT5E, PRF1, and TNF) and constructed the diagnosis model of HF.
Finally, based on the diagnosis-related genes, we constructed the miRNA-mRNA interaction network and the mRNA-TF interaction network, respectively. miRNA is a small noncoding RNA molecule with a length of about 22 nucleotides that regulates gene translation by silencing or degrading the target mRNA. They are involved in many biological processes, including differentiation and proliferation, metabolism, hemostasis, apoptosis or inflammation, and the pathophysiology of many diseases.
Overall, although some of the methods used in this study have been reported in the literature, our integration and optimization of these approaches provide new insights into heart failure (HF) research. First, this study innovatively combines DAE clustering with GSVA and immune infiltration analysis, enabling precise identification of HF subtypes. The superior performance of the DAE algorithm, validated through multiple metrics, achieves a level of comprehensive efficacy that current standard tools cannot match. Second, we developed a diagnostic model based on LASSO and SVM-RFE, and the reliability of the diagnostic genes was validated through qRT-PCR experiments. The AUC values in the training and test sets reached 0.995 and 0.95, respectively, significantly outperforming previous studies. These results suggest that the diagnostic model proposed in this study holds high clinical translational potential. Furthermore, the miRNA-mRNA and miRNA-TF regulatory networks constructed in this study provide an in-depth exploration of the molecular regulatory mechanisms of HF and, when combined with the nomogram model, demonstrate enhanced clinical application value. Compared with previous studies that rely on single tools, the systematic integration methodology and biological validation employed here enrich HF biomarker research from multiple perspectives, showcasing the uniqueness and irreplaceability of this work. Although many of the methods used in this study have been previously reported, the innovation lies in their integration and optimization, as well as their specific application in HF research.
Conclusion
This is a study to identify HF subtypes based on immunogenic cell death related genes and through multiple advanced deep-learning techniques. The two subtypes have significant differences in immunological characteristics and physiological functions. In addition, a robust heart failure diagnosis model was constructed based on machine learning models. Biomarker genes including ATG5, CASP1, CD8A, ENTPD1, IL1R1, IL10, IL17RA, MYD88, NT5E, PRF1, and TNF were identified. Finally, the interplay between biomarker genes, miRNAs, and transcription factors was explored by constructing a nomogram model. In conclusion, this article demonstrates the potential diagnostic utility of genes associated with immunogenic cell death in HF and hopes to help improve the risk stratification of HF and provide potential therapeutic targets.
Data availability
The data used in the paper was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/).
References
Krittanawong, C. et al. Clinical Update in Heart Failure with Preserved Ejection Fraction. Curr. Heart Fail. Rep. 21(5), 461–484 (2024).
Ostrominski, J. W. et al. Contemporary American and European Guidelines for Heart Failure Management: JACC: Heart Failure Guideline Comparison. JACC Heart Fail 12(5), 810–825 (2024).
Greene, S. J. et al. Worsening Heart Failure: Nomenclature, Epidemiology, and Future Directions: JACC Review Topic of the Week. J. Am. Coll Cardiol. 81(4), 413–424 (2023).
Abdin, A. et al. ‘Time is prognosis’ in heart failure: time-to-treatment initiation as a modifiable risk factor. ESC Heart Fail. 8(6), 4444–4453 (2021).
Pang, S. et al. Exploring the potential causal relationship between gut microbiota and heart failure: A two-sample mendelian randomization study combined with the geo database. Curr. Probl. Cardiol. 49(2), 102235 (2024).
Rai, A. et al. Adaptive immune disorders in hypertension and heart failure: focusing on T-cell subset activation and clinical implications. J. Hypertens 38(10), 1878–1889 (2020).
Dick, S. A. & Epelman, S. Chronic Heart Failure and Inflammation: What Do We Really Know?. Circ. Res. 119(1), 159–176 (2016).
Wang, Z. et al. Identification of key biomarkers associated with immunogenic cell death and their regulatory mechanisms in severe acute pancreatitis Based on WGCNA and machine learning. Int. J. Mol. Sci. 24(3), 3033. https://doi.org/10.3390/ijms24033033 (2023).
Zhu, M. et al. Immunogenic Cell Death Induction by Ionizing Radiation. Front. Immunol. 12, 705361 (2021).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53(D1), D672-d677 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28(11), 1947–1951 (2019).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Wang, X. et al. An immunogenic cell death-related gene expression signature in predicting prognosis of pancreatic ductal adenocarcinoma. BMC Genomics 25(1), 205 (2024).
Rusconi, C. et al. Congestive heart failure due to rapid right ventricular obliteration by metastatic malignant melanoma. Minerva Cardioangiol 44(3), 123–125 (1996).
Tesolin, M. et al. Cardiac metastases from melanoma. Radiographics 25(1), 249–253 (2005).
Wang, C. Y. et al. Cardiovascular disease and malignant melanoma. Melanoma Res. 32(3), 135–141 (2022).
Sabbah, H. N. & Sharov, V. G. Apoptosis in heart failure. Prog. Cardiovasc. Dis. 40(6), 549–562 (1998).
Chen, Q. M. & Tu, V. C. Apoptosis and heart failure: mechanisms and therapeutic implications. Am. J. Cardiovasc. Drugs 2(1), 43–57 (2002).
Liao, M. et al. Main active components of Si-Miao-Yong-An decoction (SMYAD) attenuate autophagy and apoptosis via the PDE5A-AKT and TLR4-NOX4 pathways in isoproterenol (ISO)-induced heart failure models. Pharmacol. Res. 176, 106077 (2022).
Booz, G. W., Day, J. N. & Baker, K. M. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J. Mol. Cell. Cardiol. 34(11), 1443–1453 (2002).
Okonko, D. O. et al. Erythropoietin resistance contributes to anaemia in chronic heart failure and relates to aberrant JAK-STAT signal transduction. Int. J. Cardiol. 164(3), 359–364 (2013).
Du, J., Liu, Y. & Fu, J. Autophagy and Heart failure. Adv. Exp. Med. Biol. 1207, 223–227 (2020).
Gao, G. et al. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int. J. Mol. Med. 45(1), 195–209 (2020).
Shirazi, L. F. et al. Role of inflammation in Heart failure. Curr. Atheroscler. Rep 19(6), 27 (2017).
Schiattarella, G. G., Rodolico, D. & Hill, J. A. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc Res. 117(2), 423–434 (2021).
Triposkiadis, F. et al. Obesity, inflammation, and heart failure: links and misconceptions. Heart Fail. Rev. 27(2), 407–418 (2022).
Levine, B. et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 323(4), 236–241 (1990).
Müller-Ehmsen, J. & Schwinger, R. H. TNF and congestive heart failure: therapeutic possibilities. Expert Opin. Ther. Targets 8(3), 203–209 (2004).
Ueland, T. et al. TNF revisited: osteoprotegerin and TNF-related molecules in heart failure. Curr. Heart Fail Rep. 9(2), 92–100 (2012).
Tijsen, A. J. et al. MiR423-5p as a circulating biomarker for heart failure. Circ. Res. 106(6), 1035–1039 (2010).
Vilella-Figuerola, A. et al. Systems biology in chronic heart failure-identification of potential miRNA regulators. Int. J. Mol. Sci. 23(23), 15226. https://doi.org/10.3390/ijms232315226 (2022).
Deng, J. & Zhong, Q. Advanced research on the microRNA mechanism in heart failure. Int. J. Cardiol 220, 61–64 (2016).
Funding
The biological experiments involved in this paper (qRT-PCR) were performed by the first author at his own expense. The cell lines used were commercially purchased.
Author information
Authors and Affiliations
Contributions
Zhihui Ma: Conceptualization, Methodology, Software, Visualization, Writing – Original Draft Preparation, Writing-review & editing. Shixin Ma: Conceptualization, Methodology, Software, Visualization,Writing-review & editing. Bin Chen: Conceptualization, Methodology, Software, Writing-review & editing. Yongjun Zhang, and Jinmei Zeng: Data curation, Software, Writing-review & editing. Jianping Tao: Visualization, Writing-review & editing. Yu Hu: Data curation, Writing-review & editing. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, Z., Ma, S., Chen, B. et al. Classification patterns identification of immunogenic cell death-related genes in heart failure based on deep learning. Sci Rep 15, 5633 (2025). https://doi.org/10.1038/s41598-025-89333-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-89333-1
