
Abstract
The Ryukyu archipelago is located southwest of the Japanese islands, and people originally from this region, the Ryukyu population, have a unique genetic background distinct from that of other populations, including people from mainland Japan. However, few genetic studies have focused on the Ryukyu population. In this study, we performed genome-wide association studies (GWAS) on the serum levels of alanine aminotransferase (ALT, n = 15,224), aspartate aminotransferase (AST, n = 15,203), and gamma-glutamyl transferase (GGT, n = 14,496) in the Ryukyu population. We found 13 loci with a genome-wide significant association (P < 5 × 10−8), three for ALT, four for AST, and six for GGT, including one novel locus associated with ALT: rs117595134-A in HMMR/HMMR-AS1, ß = − 0.131, standard error = 0.024, P = 4.90 × 10−8. Rs117595134-A is common in the Japanese population but is not observed in other ethnic populations in the 1000 genomes database. Additionally, 77 of 80 loci derived from Korean GWAS and 541 of 716 loci from European GWAS showed the same directions of effect (P = 1.41 × 10−19, P = 2.50 × 10−44, binomial test), indicating that most of susceptibility loci are shared between the Ryukyu population and other ethnic populations.
Similar content being viewed by others


Introduction
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma-glutamyl transferase (GGT) levels are widely used in clinical tests to evaluate liver injury or function. The hepatocellular or cholestatic injury, caused by various conditions, including hepatitis virus infection, malignancy, autoimmune diseases, alcohol intake, obesity, and drugs, leads to the increase of the levels of these serum liver enzymes1,2. Although AST and ALT are abundant in the canalicular membrane of hepatocytes, the enzymes also exist in extrahepatic tissues such as cardiac muscle, skeletal muscle, kidney, and brain, especially AST. Therefore, serum AST levels are also elevated in individuals with myocardial infarction or myositis. In contrast, because the amount of ALT is lower in other tissues, ALT elevation is relatively specific to hepatocellular injury. Elevated serum GGT levels usually reflect bile stasis, but serum GGT levels are also elevated during the detoxification of xenobiotics, which include alcohol3 or in patients treated with enzyme-inducing anticonvulsant drugs4.
Additionally, several genetic variants have been shown to abolish these enzyme activities and significantly affect the results of clinical measurements of liver enzymes5,6, because the serum levels of these enzymes are usually determined by measuring their enzyme activities in clinical practice. Therefore, it is expected that the serum liver enzyme levels are affected by genetic factors related or unrelated to liver injury5,6,7.
During the last two decades, genome-wide association studies (GWAS) have been extensively performed worldwide, and a substantial number of loci were successfully identified to be associated with common diseases and quantitative traits8,9,10. Large-scale GWAS for serum liver enzyme levels have also been performed, which identified 954 loci in European11,12, 521 loci in trans-ancestry13, and 284 loci in Japanese studies10.
The Ryukyu Archipelago is located southwest of the Japanese Islands and comprises dozens of islands. People from the Ryukyu Archipelago have been shown to possess unique genetic backgrounds that differ from those of the mainland Japan14,15,16,17,18. However, no GWAS has been conducted specifically on the Ryukyu population. Because GWAS using genetically isolated populations identified novel loci with larger effect sizes, even in a smaller sample size19,20, GWAS focused on the Ryukyu population is important and necessary to elucidate the genetic factors not only specific to the Ryukyu population but also for other populations.
In this study, we conducted a GWAS for serum liver enzyme levels using the Ryukyu population for the first time from two independent Japanese study groups: Okinawa Bioinformation Bank (OBi)16,17,21 and BioBank Japan (BBJ)22,23,24.
Results
Population structure analyses
The overall study design is shown in Supplementary Fig. 1. We analyzed 10,285 individuals registered in the OBi using principal component analysis (PCA), along with individuals in the 1KGP phase 3 reference panel (AFR, n = 661; AMR, n = 347; EAS, n = 504; EUR, n = 503; SAS, n = 489), and defined 10,136 individuals as the Japanese population (Supplementary Fig. 2b). Subsequent PCA combined with uniform manifold approximation and projection (UMAP) for the Japanese population revealed that the population was further divided into five clusters (Supplementary Fig. 2c). Information on the birthplace of grandparents for each participant indicated that one cluster consisted of people from mainland Japan, the Hondo cluster (OBi Hondo, n = 1421), and the remaining four consisted of people from the Ryukyu archipelago, the Ryukyu cluster (OBi Ryukyu, n = 8715) (Supplementary Fig. 2c).
We also performed PCA on 184,092 participants registered in BBJ and 1KGP Phase 3 and removed 100 individuals defined as outliers not belonging to JPT or CHB (Supplementary Fig. 2d). From the PCA results of the remaining 183,992 individuals in BBJ, we defined two clusters: BBJ Ryukyu and BBJ Hondo (Supplementary Fig. 2e).
GWAS for serum liver enzyme levels in the Ryukyu population
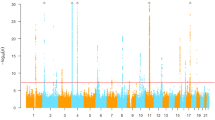
The clinical characteristics of the Ryukyu population brought to GWAS are presented in Table 1. We observed 13 independent loci with GWAS (P < 5 × 10−8) for three serum liver enzyme traits (Fig. 1, Table 2, Supplementary Tables 1, 2, 3), three for ALT (n = 15,224), rs3747207 in PNPLA3, rs3782886 in BRAP, and rs117595134 in HMMR/HMMR-AS1, four for AST (n = 15,203), rs76850691 in GOT1, rs3747207 in PNPLA3, rs369827206 in MRC1, rs147992802 near NAA25, and six for GGT, (n = 14,496), rs4049918 in GGT1, rs5760109 near MIF-AS1, rs58367757 in HNF1A, rs7678352 in ZNF827, rs11066325 in PTPN11, rs7519043 near EPHA2 (Fig. 2, Supplementary Fig. 3). Most of these loci have been previously reported to be associated with serum liver enzyme levels, except for the HMMR/HMMR-AS1 locus, whose association with ALT or other liver enzyme traits has not been previously reported.

Results of meta-analyses for genome-wide association studies for serum liver enzyme levels in the Ryukyu population. Manhattan plots (left) and Quantile—Quantile plots (right) of GWAS for serum (a) ALT, (b) AST, and (c) GGT levels. Red lines indicate the threshold for the genome-wide significance level (P = 5 × 10−8). Green plots correspond to variants that attained genome-wide significance. A novel locus is shown by red letters.

A regional plot for association of HMMR/MMMR-AS1 locus with serum ALT values in the Ryukyu population. The lead variant is shown as purple diamond. The other variants are colored according to the extent of LD estimated in 3256 Japanese WGS samples from the BBJ and 2504 individuals from the 1KGP phase 3.
The minor A-allele of rs117595134 within HMMR/HMMR-AS1 was observed only in the Japanese in the 1000 genomes database, 5.3% in JPT (Supplementary Table 4). In this study, the A-allele frequencies were significantly higher in the Ryukyu population than in the Hondo population (P = 2.59 × 10−6, 6.0%, 6.0%, 2.9%, and 2.8% in OBi Ryukyu, BBJ Ryukyu, OBi Hondo, and BBJ Hondo, respectively; Table 3). Additionally, the absolute values of effect sizes (ß) of the A-allele are also significantly larger in the Ryukyu population than in the Hondo population (P for heterogeneity = 2.16 × 10−4, ß = – 0.133, −0.128, 0.015, −0.012 in OBi Ryukyu, BBJ Ryukyu, OBi Hondo, and BBJ Hondo, respectively, Table 3).
Replication study for previously reported loci in the Ryukyu population
We conducted replication studies of the loci reported in the Korean Genome and Epidemiology Study (KoGES)25 and UK Biobank11,12. Among the 24 loci associated with ALT in 126 K Korean individuals in KoGES, two loci showed genome-wide significant association (P < 5.00 × 10−8), and nine loci showed a significant association in the Ryukyu population (P < 2.08 × 10−3 = 0.05/24). Similarly, three out of 25 loci for AST and four out of 43 loci for GGT showed genome-wide significant association and eight loci for AST and 13 loci for GGT showed significant associations in the Ryukyu population (P < 2.00 × 10−3 = 0.05/25, P < 1.16 × 10−3 = 0.05/43). In total, 77 of 80 signals (20 of 22 in ALT, 22 of 22 in AST, and 35 of 36 in GGT) showed the same directions of effect (P = 1.21 × 10−4 for ALT, P = 4.77 × 10−7 for AST, and P = 1.08 × 10−9 for GGT, P = 1.41 × 10−19 in total, binomial test) (Supplementary Table 5).
Among the 247 loci associated with ALT in 437 K European individuals in the UK Biobank, one locus showed a genome-wide significant association, and four loci showed a significant association in the Ryukyu population (P < 2.02 × 10−4 = 0.05/247). Regarding European-derived loci for AST in 388 K Europeans and GGT in 437 K Europeans, one locus for AST and 5 loci for GGT showed genome-wide significant association, and 3 of 336 loci for AST and 12 of 371 loci for GGT showed significant association in the Ryukyu population (P < 1.49 × 10−4 = 0.05/336 for AST, P < 1.35 × 10−4 = 0.05/371 for GGT) (Supplementary Tables 6, 7, 8). In total, 541 of 716 signals (142 of 185 in ALT, 174 of 249 in AST, and 225 of 282 in GGT) showed the same directions of effect (P = 1.47 × 10−13 for ALT, P = 3.11 × 10−10 for AST, and P = 8.94 × 10−25 for GGT, P = 2.50 × 10−44 in total, binomial test).
Gene-based analyses using MAGMA
Next, we performed gene-based association analyses using MAGMA using data from the GWAS meta-analyses in the Ryukyu population and identified 22 genes significantly associated with liver enzyme traits: five for ALT, three for AST, and 14 for GGT (P < 2.63 × 10−6 = 0.05/18,980, Supplementary Tables 9, 10, 11, Supplementary Fig. 4). These genes are located within ± 500 kb of the individual loci that have been previously reported in GWAS for serum liver enzyme levels. Of these, the association of two genes, CHCHD10 and C22orf15, with GGT, has not been previously reported by gene-based analyses. We conducted gene-set analyses using MAGMA but could not identify any curated gene sets or Gene Ontology terms significantly associated with ALT, AST, and GGT (Supplementary Tables 12, 13, 14). Tissue expression enrichment analyses using MAGMA revealed that the genes for GGT, identified through gene-based analysis, were significantly enriched in the liver tissue. In contrast, no significant enrichment was observed for genes associated with ALT and AST (Supplementary Tables 15, 16, 17, Supplementary Fig. 5).
Discussion
In this study, we conducted GWAS meta-analyses using the Ryukyu population and found 13 loci with genome-wide significant associations for three serum liver enzyme traits, one novel locus, HMMR/HMMR-AS1 for ALT, and 12 known loci.
GWAS have been extensively performed to identify the genetic loci associated with common diseases or quantitative traits, such as laboratory tests. Thousands of genetic loci have been reported to be associated with serum liver enzyme levels, including ALT, AST, and GGT, through GWAS using nearly a million participants, mostly individuals of European origin. A large-scale GWAS for the liver enzyme traits in the Japanese has also been performed, and additional 130 loci associated with ALT, AST, and GGT have been identified10,23,24.
In this study, we performed GWAS for serum liver enzyme levels, focusing on the Ryukyu population for the first time. Although the Japanese population is considered genetically homogenous, it has been known that there are two genetically distinct groups, Hondo for the mainland Japan and Ryukyu for the Ryukyu archipelago located in the south-western part of Japanese islands, from the results of principal component analyses using information of genome-wide single nucleotide polymorphism genotypes14,15. Additionally, it has been reported that people from the Ryukyu archipelago are further divided into several sub-groups16,17. Therefore, the Ryukyu population has a unique genetic background that differs from other regions. Since GWAS for isolated populations with unique genetic backgrounds have been reported to be useful for identifying novel loci with larger effect sizes even in smaller sample sizes, such as TBC1D4 for type 2 diabetes in Greenlandic Inuits19 or CREBRF for body mass index in Samoans20, it is suggested that GWAS using populations with unique genetic backgrounds, such as the Ryukyu population, is useful and important for identifying additional novel loci even if the sample size is not very large.
Our GWAS meta-analysis of ALT identified a novel SNP locus, rs117595134, within HMMR/HMMR-AS1 on chromosome 5. In this study, the A-allele frequencies are significantly higher in the Ryukyu population than in the Hondo population, and the absolute values of effect sizes (ß) of the A-allele are also significantly larger in the Ryukyu population than in the Hondo population. Therefore, the higher allele frequency and larger effect size for this variant observed in the Ryukyu population indicate that HMMR/HMMR-AS1 is likely to be a Ryukyu population-specific locus associated with ALT measures. We further searched for the whole genome sequencing data from 254 individuals belonging to the Ryukyu population to identify functional variants with high linkage disequilibrium to rs117595134 (r2 ≥ 0.6), but no variant exists within ± 5 Mb from rs117595134, showing a significant deleterious effect evaluated using Combined Annotation Dependent Depletion software (Supplementary Table 18).
This variant is located in the intron of HMMR and HMMR-AS1. HMMR encodes a hyaluronan-mediated motility receptor, also known as CD168 or RHAMM26. The HMMR has been reported to be overexpressed in various tumor tissues, such as breast cancer27 and gastrointestinal cancer28 and implicated in promoting carcinogenesis29. Additionally, HMMR was recently reported as a critical gene associated with both nonalcoholic steatohepatitis and hepatocellular carcinoma (HCC), and silencing HMMR reduces HCC cell proliferation and tumor growth while also regulating cell cycle progression30,31. However, no evidence suggests a role for HMMR in directly or indirectly regulating serum ALT levels. Further studies are required to elucidate the molecular mechanisms through which this locus regulates serum ALT levels.
This study has several limitations. First, although this was the largest GWAS of serum liver enzyme traits in the Ryukyu population, the sample size was insufficient. Therefore, further studies are required to validate the association of the novel locus. Since statistical power to detect significant association of rs117595134 with ALT value in BBJ-Hondo was insufficient (14.4% for α = 0.05, Supplementary Table 19), additional replication sets with larger sample size or with larger effect size would be required to validate the association of this locus. Second, precise information on liver diseases, such as a history of hepatitis B or C viral infection, presence of diseases including metabolic dysfunction-associated fatty liver disease and HCC, is lacking in the OBi participants. Third, differences in the characteristics of participants between OBi and BBJ exist. BBJ is a hospital-based disease cohort, whereas OBi is a population-based study, and the mean age of BBJ participants is higher than that of OBi participants. These differences may have reduced the statistical power of the meta-analysis. Fourth, some databases for in silico analyses have been built from European data, which reduces the statistical power of these analyses.
In conclusion, we performed a GWAS for serum liver enzyme levels in the Ryukyu population for the first time and found several independent loci with genome-wide significant associations, including a novel locus associated with serum ALT. The lead variant located in the intron of HMMR/HMMR-AS1, rs117595134, is specific to the Japanese population, especially the Ryukyu population. Further studies are required to elucidate the biological mechanisms of this locus that contribute to the regulation of serum ALT levels.
Methods
Participants, genotyping, and imputation
We analyzed individuals registered in Okinawa Bioinformation Bank (OBi), which is a population-based study in Okinawa Prefecture established in 201616,17,21. Individual’s values for laboratory tests, including serum ALT, serum AST, and serum GGT; anthropometric measurements, including height, body weight, and body mass index; and lifestyle questionnaire information, including alcohol consumption and smoking status, were obtained from results of health check-up. Genomic DNA was extracted from peripheral leukocytes or saliva using a standard procedure, and SNP genotyping was performed using the Infinium Asian Screening Array v1.0 BeadChip (Illumina, Inc. CA, USA).
We also analyzed participants registered in BioBank Japan (BBJ), a hospital-based cohort study consisting of patients with 47 diseases from 12 medical institutions in Japan22,23,24. Clinical information for laboratory test results, including serum ALT, AST, and GGT; anthropometric measures, including height, body weight, and body mass index; lifestyle questionnaire, including alcohol drinking behavior; and the status of 47 target diseases were obtained from the clinical records. Individuals with liver diseases, including liver cirrhosis, Hepatitis B or C virus infection, hepatocellular carcinoma, gallbladder carcinoma, or cholangiocarcinoma were excluded from the analyses. Genotyping was performed using Illumina Infinium Omni Express, Human Exome, Infinium Omni Express Exome v1.0, or Infinium Omni Express Exome v1.2 (Illumina Inc, CA, USA).
As a quality control (QC), we removed individuals (i) other than Japanese (outliers determined using PCA with 1KGP phase 3 as a reference panel), (ii) sex mismatch between clinical and genotypic information, (iii) sample call rate < 0.98, and (iv) one of the pairs of identical twins or duplicate samples determined using identity-by-descent (PIHAT > 0.95). For QC of SNPs, we excluded SNPs with (i) SNP call rate < 0.98, (ii) deviated genotype distribution from Hardy Weinberg Equilibrium (P ≤ 1.0 × 10−6), (iii) minor allele frequency < 0.01. The QC process was performed using PLINK v1.9.
The genotyped data were phased using Eagle (version 2.3) and imputed using linkage disequilibrium (LD) information for 3,256 Japanese WGS from the BBJ and 2,504 individuals from the 1KGP (phase3v5)32 using Minimac4 software. We excluded variants with low imputation quality (Rsq < 0.7) and low minor allele frequency (minor allele frequency < 0.005).
GWAS and GWAS meta-analyses
To perform GWAS for the values of serum liver enzymes, we standardized serum ALT, AST, and GGT values using the following procedures: (i) we first transformed individual values to common logarithms, (ii) distributions of log-transformed values were evaluated in females and males separately, and samples with above 4SD or below − 4SD were removed, (iii) linear regression analysis was performed by adjusting age, sex, principal components 1 to 10, drinking status (none; 0, sometimes; 1, almost every day; 2 in OBi, no; 0 or yes; 1 in BBJ) and the each disease statuses of the 42 diseases (unaffected;0, affected;1 only for BBJ), and (iv) the residuals for the regression analyses were z-standardized and applied to GWAS as quantitative values (Supplementary Fig. 6). We conducted GWAS for serum liver enzyme levels using a linear mixed model in BOLT-LMM (v.2.3.6)33, which effectively accounts for population stratification and cryptic relatedness, by assuming candidate SNP is modeled as a fixed effect, and environmental effect and polygenic effect by residual SNPs other than tested SNP are modeled as random effects.
We performed meta-analyses using the inverse-variance weighted method with a fixed-effects model and assessed heterogeneity using Cochran’s Q-test with METAL software (v.2020-05-05)34. The genomic inflation factors (λGC) of GWAS meta-analyses in the Ryukyu population for the ALT, AST, and GGT were 1.03, 1.04, and 1.06 respectively (Fig. 1), and those of the LDSC intercepts were 1.0109, 1.0097, and 1.0398 respectively (Supplementary Table 20). These results indicate there was no significant inflation attributable to population stratification and other confounding factors. We defined independent genome-wide significant loci as genomic positions within ± 500 kb from the lead variant. We searched for the genome-wide significant loci of this study in the GWAS catalog, and if there are no variants reported as associated with each serum liver enzyme level in the locus within ± 1 Mb from the lead variants of this study, we determined these loci as novel loci associated with the liver enzyme trait. We annotated rsIDs in the dbSNP database, the closest genes to the lead variants, using ANNOVAR software35.
LD score regression
We conducted LD Score Regression v1.0.136 using GWAS meta-analysis data for ALT, AST and GGT in the Ryukyu population. For the regression, we used the East Asian LD scores and summary statistics of high-quality common SNPs present in the HapMap 3 reference panel for each trait.
Gene-based and gene-set association analyses
Gene-based and gene-set association analyses were conducted using MAGMA v1.08, as implemented in FUMA v1.6.137. We used summary statistics data for the meta-analysis in this study. These included 8,413,895 variants for ALT, 8,412,728 for AST, and 8,395,794 for GGT. The variant information was converted from GRCh38 (hg38) to GRCh37 (hg19) using the lift-over method (LiftOver; https://genome.sph.umich.edu/wiki/LiftOver). Individual variants were mapped to 18,980 protein-coding genes in the hg19 build, and gene-based p-values were calculated. Information on LD from the 1KGP phase 3 EAS was used, and window sizes were set to range from 2 kb upstream to 1 kb downstream of the genes. The statistical significance threshold for the gene-based analysis was set at a Bonferroni-corrected P-value, < 2.63 × 10−6 (0.05/18,980). In the gene-set analysis, 17,009 gene sets (curated gene sets and Gene Ontology terms) from MsigDB v7.0 were assessed, and the significance threshold was set at P < 2.94 × 10−6 (0.05/17,009). We conducted tissue expression enrichment analyses using the MAGMA tool and RNA sequencing data from 54 different tissue types sourced from the GTEx v8.
In silico functional annotation
The functional category was annotated using Combined Annotation Dependent Depletion (CADD) v1.638 to evaluate variant pathogenicity.
Statistical power calculation
Power calculations for the association of rs117595134 within novel locus were given for a range of allele frequencies and effect sizes based on the additive genetic model and using the Quanto (Version 1.2.3) program39.
Softwares
Manhattan and quantile–quantile plots were drawn using R software version 4.0.5 [https://CRAN.R-project.org/], and regional association plots were generated using LocusZoom40.
Data availability
The summary statistics of GWAS meta-analyses for AST, ALT, GGT in the Ryukyu population are available from Database Center for Life Science (DBCLS) website (https://humandbs.dbcls.jp/en/), ID hum0496.
References
Newsome, P. N. et al. Guidelines on the management of abnormal liver blood tests. Gut 67, 6–19 (2018).
Umashankar, R. Continuing medical education: January 2017: ACG clinical guideline: Evaluation of abnormal liver chemistries. Am. J. Gastroenterol. 112, 17 (2017).
Singh, M., Gupta, S., Singhal, U., Pandey, R. & Aggarwal, S. K. Evaluation of the oxidative stress in chronic alcoholics. J. Clin. Diagn. Res. 7, 1568–1571 (2013).
Tutor-Crespo, M., Hermida, J. & Tutor, C. Isoforms of serum gamma-glutamyltransferase in epileptic patients treated with enzyme-inducing anticonvulsant drugs. J. Pharm. Pharm. Sci. 7, 353–358 (2004).
Kulecka, M. et al. A heterozygous mutation in GOT1 is associated with familial macro-aspartate aminotransferase. J. Hepatol. 67, 1026–1030 (2017).
Darin, N. et al. γ-Glutamyl transpeptidase deficiency caused by a large homozygous intragenic deletion in GGT1. Eur. J. Hum. Genet. 26, 808–817 (2018).
Caon, E. et al. Exploring the impact of the PNPLA3 I148M variant on primary human hepatic stellate cells using 3D extracellular matrix models. J. Hepatol. 80, 941–956 (2024).
Chen, V. L. et al. Genome-wide association study of serum liver enzymes implicates diverse metabolic and liver pathology. Nat. Commun. 12, 816 (2021).
Chen, C.-Y. et al. Analysis across Taiwan Biobank, Biobank Japan, and UK Biobank identifies hundreds of novel loci for 36 quantitative traits. Cell Genom. 3, 100436 (2023).
Koyama, S. et al. Population-specific putative causal variants shape quantitative traits. Nat. Genet. 56, 2027–2035 (2024).
Pazoki, R. et al. Genetic analysis in European ancestry individuals identifies 517 loci associated with liver enzymes. Nat. Commun. 12, 2579 (2021).
Gao, C. et al. Genome-wide association analysis of serum alanine and aspartate aminotransferase, and the modifying effects of BMI in 388k European individuals. Genet. Epidemiol. 45, 664–681 (2021).
Ward, L. D. et al. GWAS of serum ALT and AST reveals an association of SLC30A10 Thr95Ile with hypermanganesemia symptoms. Nat. Commun. 12, 4571 (2021).
Yamaguchi-Kabata, Y. et al. Japanese population structure, based on SNP genotypes from 7003 individuals compared to other ethnic groups: effects on population-based association studies. Am. J. Hum. Genet. 83, 445–456 (2008).
Sakaue, S. et al. Dimensionality reduction reveals fine-scale structure in the Japanese population with consequences for polygenic risk prediction. Nat. Commun. 11, 1569 (2020).
Matsunami, M. et al. Fine-scale genetic structure and demographic history in the Miyako Islands of the Ryukyu Archipelago. Mol. Biol. Evol. 38, 2045–2056 (2021).
Liu, X. et al. Natural selection signatures in the Hondo and Ryukyu Japanese subpopulations. Mol. Biol. Evol. 40, msad231 (2023).
Liu, X. et al. Decoding triancestral origins, archaic introgression, and natural selection in the Japanese population by whole-genome sequencing. Sci. Adv. https://doi.org/10.1126/sciadv.adi8419 (2024).
Moltke, I. et al. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 512, 190–193 (2014).
Minster, R. L. et al. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat. Genet. 48, 1049–1054 (2016).
Matsunami, M. et al. Genome-wide association studies for pelvic organ prolapse in the Japanese population. Commun. Biol. 7, 1188 (2024).
Nagai, A. et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Kanai, M. et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 50, 390–400 (2018).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
Kim, Y. J. et al. The contribution of common and rare genetic variants to variation in metabolic traits in 288,137 East Asians. Nat. Commun. 13, 6642 (2022).
Maxwell, C. A. et al. RHAMM is a centrosomal protein that interacts with dynein and maintains spindle pole stability. Mol. Biol. Cell 14, 2262–2276 (2003).
Tarullo, S. E. et al. Receptor for hyaluronan-mediated motility (RHAMM) defines an invasive niche associated with tumor progression and predicts poor outcomes in breast cancer patients. J. Pathol. 260, 289–303 (2023).
Koelzer, V. H. et al. Expression of the hyaluronan-mediated motility receptor RHAMM in tumor budding cells identifies aggressive colorectal cancers. Hum. Pathol. 46, 1573–1581 (2015).
Yang, C., Li, C., Zhang, P., Wu, W. & Jiang, X. Redox responsive hyaluronic acid nanogels for treating RHAMM (CD168) over-expressive cancer, both primary and metastatic tumors. Theranostics 7, 1719–1734 (2017).
Zhang, D. et al. Oleate acid-stimulated HMMR expression by CEBPα is associated with nonalcoholic steatohepatitis and hepatocellular carcinoma. Int. J. Biol. Sci. 16, 2812–2827 (2020).
Meng, Q., Li, X. & Xiong, X. Identification of hub genes associated with non-alcoholic steatohepatitis using integrated bioinformatics analysis. Front. Genet. 13, 872518 (2022).
Flanagan, J. et al. Population-specific reference panel improves imputation quality for genome-wide association studies conducted on the Japanese population. Commun. Biol. 7, 1665 (2024).
Loh, P.-R. et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 47, 284–290 (2015).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Rentzsch, P., Schubach, M., Shendure, J. & Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 13, 31 (2021).
Gauderman, W. J. Sample size requirements for matched case-control studies of gene-environment interaction. Stat. Med. 21, 35–50 (2002).
Pruim, R. J. et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337 (2010).
Acknowledgements
We thank all participants in the Okinawa Bioinformation Bank (OBi) Project, and BioBank Japan (BBJ) Project. We would like to express our gratitude to the staff at all study sites for their assistance in sample collection. This work was partly supported by a grant from the promotion project of innovation-eco system collaborating organization from the Okinawa prefecture to SM, and a grant from the advanced medical research project from University of the Ryukyus to MM.
Author information
Authors and Affiliations
Consortia
Contributions
Conception and Design: N.O., M.I., S.M. Resources: OBi; S.M., BBJ: C.T., K.M. Data curation: N.O., M.M., X.L.. Analysis – Genome-wide association studies: N.O., M.M. Analysis – QC and population structure analysis: N.O., M.M., X.L., A.Y., A.J. Analysis – Gene based, gene set analyses: N.O. Analysis – Haplotype phasing in the Ryukyu population: R.K. Supervising and data interpretation: M.M., M.I., C.T., S.M. Writing – original draft: N.O. Writing – review & editing: M.M., M.I., C.T., S.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declarations
All the participants provided written informed consent to participate in this study. The study protocol conformed to the provisions of the Declaration of Helsinki and was approved by the Ethics Committee of the University of the Ryukyus (Nishihara, Japan) for Medical and Health Research Involving Human Subjects (Approval No. 21-1826-08-02-00, 18-2300-05-00-00), the Ethical Committee at the Institute of Medical Sciences, The University of Tokyo (Tokyo, Japan) (Approval No. 2022-61-0119), and the RIKEN Center for Integrative Medical Sciences (Yokohama, Japan) (Approval No. RIKEN-Y-2022-068).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ohyama, N., Matsunami, M., Imamura, M. et al. A variant in HMMR/HMMR-AS1 is associated with serum alanine aminotransferase levels in the Ryukyu population. Sci Rep 15, 6494 (2025). https://doi.org/10.1038/s41598-025-90195-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-90195-w
