
Abstract
Although there is some evidence of an association between Alzheimer’s disease polygenic risk score (AD PRS) and cognitive function, limited validations have been performed in large populations. We investigated the relationship between AD PRS and cognitive function in the UK Biobank in over 276,000 participants and further validated the association in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) sample. We developed the AD PRS (excluded the APOE variants) in the middle age UK Biobank participants (age ranged 39–72, mean age 57 years) of European ancestries by LDpred2. To validate the association of AD PRS and cognitive function internally in the UK Biobank, we linearly regressed standardized cognitive function on continuous standardized AD PRS with age at cognitive test, sex, genotyping array, first 10 principal components of genotyping, smoking, education in years, body mass index, and apolipoprotein E gene ε4 (APOE4) risk allele dosages. To validate the associations externally, we ran the linear mixed effects model in the ADNI sample free of dementia (age ranged 55–91, mean age 73), including similar covariates as fixed effects and participants’ IDs as the random effect. Stratification by age, APOE4 carrier status, and cognitive status (cognitively normal or mild cognitive impairment) was also investigated. Our study validated the associations of AD PRS and cognitive function in both midlife and late-life observational cohorts. Although not all of the cognitive measures were significantly associated with AD PRS, non-verbal fluid reasoning (matrix pattern completion, β = − 0.022, P = 0.003), processing speed (such as symbol digit substitution, β = − 0.017, P = 1.08E−05), short-term memory and attention (such as pairs matching, β = − 0.014, P = 1.66E−10), and reaction time (β = − 0.010, P = 1.19E−06) were inversely associated with increasing AD PRS in the UK Biobank. Higher likelihood of cognitive impairment was also associated with higher AD PRS in the ADNI cognitive normal individuals (AD assessment scale β = 0.079, P = 0.02). In summary, we confirmed that poorer cognitive function was associated with a higher polygenic AD risk, and suggested the potential utility of the AD PRS in identifying those who may be at risk for further cognitive decline.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD), which accounts for 60–70% of dementia, is a common polygenic progressive neurodegenerative disorder mainly affecting people in late life. It is one of the major causes of disability and dependency1,2. Although some medical management can improve the quality of life for people living with AD, and new medications may be able to slow the progression of the disease to some extent, there is no effective cure for the illness3. Screening for AD before it is clinically detectable or during the early stages of the disease is therefore crucial. Cognitive impairment based on episodic memory or global cognitive tests is typically used to identify at-risk populations4,5. However, prediction of future AD risk solely based on cognitive tests is not sufficient because there is substantial baseline variation in cognitive performance and psychosocial and medical factors may contribute to cognitive decline in the absence of latent degenerative or vascular dementia pathologies6,7.
Beyond abovementioned cognitive indicators, genetic component can also be a powerful biomarker for AD risk stratification. The overall heritability of AD is 40–70% and only a part of it captured by the single-nucleotide variants (SNVs) identified by genome-wide association studies (GWAS)8,9. A polygenic risk score (PRS), which represents the collective influence of many SNVs, can enhance the risk prediction of AD10. The exploration of AD PRS and cognitive function associations may reveal potential pathology of cognitive impairment leading to AD and may suggest the usefulness of AD PRS in identifying subtle cognitive changes in normal and/or mildly impaired populations11,12,13,14.
Both the establishment of large AD consortia15 and PRS methods development provide opportunities to characterize AD genetic risks, i.e., capture more genetic variability of AD. For example, recent GWAS have identified nearly one hundred AD risk loci12,15, and one of the most widely used PRS calculation methods “LDpred2” has shown better predictive accuracy and faster computational speed in PRS estimation than the prior version “LDpred” and other PRS methods (e.g., lassosum, PRS-CS, SBayesR) in European populations16. New findings on AD PRS and cognitive function may arise by integrating recent PRS methods with new AD GWAS results. Thus, we will utilize the summary results from the most recent AD GWAS to examine the association of AD PRS and cognitive function in the UK Biobank. We will validate associations we identify in the Alzheimer’s Disease Neuroimaging Initiative (ADNI).
Methods
Populations
We used the UK Biobank as the training set for the AD PRS derivation, as well as the internal testing set for the association of AD PRS and cognitive function. The UK Biobank is a large prospective cohort of over 500,000 participants with rich genetic and health information17,18. We initially included 339,506 unrelated middle-age participants (i.e., no kinship was found in the UK Biobank Field ID 22021) with available imputed genotypes. We then excluded 62,837 participants of non-European ancestries (Field ID 22006 not in Caucasian) and 13 individuals with prevalent AD at baseline. This left 276,656 participants in the PRS training set (Fig. 1). Those participants with cognitive tests were included as the internal testing set in the UK Biobank.

Analytic framework of AD polygenic score derivation and its association with cognitive functions. Created in https://BioRender.com.
We externally validated the AD PRS and cognitive function associations in ADNI, which is a longitudinal study focusing on early detection and disease progression with multiple time-points of data collections19. We included 685 individuals without dementia at baseline and high quality whole genome sequencing data in the external testing set from ADNI (Fig. 1).
Genetic data
Two Affymetrix (BiLEVE Axiom array and Affymetrix UK Biobank Axiom array) genotyping arrays were utilized in the UK Biobank to code over 850,000 variants18. The genotype data in the UK Biobank was imputed by the UK10K and 1000 Genomes Phase 3 reference panels for over 480,000 participants who passed the sample-based quality control (QC) including heterozygosity check, less than 5% missing rate, no sex mismatch, no duplicates, and no sample mishandling. Genetic variants with minor allele frequency (MAF) < 0.0001 were filtered out before imputation.
The whole genome sequencing was performed in 818 ADNI participants by Illumina’s non-CLIA laboratory at roughly 30–40× coverage20,21. Genetic variants with Hardy-Weinberg Equilibrium < 1e−06, and missing rate < 95% were included as passed QC variants in the PRS calculation.
Polygenic risk score derivation
We used LDpred2-grid to derive the AD PRS. The GWAS summary statistics were based on the meta-analysis of over 100,000 AD or proxy AD cases with European ancestry12. We selected the overlapped variants among AD GWAS summary statistics, UK Biobank imputed genotype data, and HapMap3+ (with over 1,400,000 SNVs across the whole genome) to calculate the PRS. Variants within the APOE linkage region (Genome Build 37, chromosome 19: 4500000–4580000) were excluded22, leaving 1,415,926 SNVs to calculate the PRS. The LD reference panel was based on genotyping 15,000 randomly selected unrelated individuals of European ancestry in the UK Biobank. The tuning hyper-parameters in the LDpred2-grid were heritability (0.0083, 0.0248, 0.0579, 0.0827, or 0.1157), proportion of causal variants (0.0001, 0.00018, 0.00032, 0.00056, 0.001, 0.0018, 0.0032, 0.0056, 0.01, 0.018, 0.032, 0.056, 0.1, 0.18, 0.32, 0.56, 1), and modeling sparsity options (yes or no). The PRS with the highest concordance statistics (C-stats) in the prediction of incident AD was considered optimal and used in the downstream analyses.
To generate the PRS in ADNI, we used the 1,387,064 SNVs both in the ADNI whole genome sequencing data and in the optimal PRS from the UK Biobank described above (covered 98% SNVs of the UK Biobank). The ADNI PRS was then calculated as a linear combination of those variants and their shrinkage effect sizes derived from the optimal PRS from UK Biobank.
AD diagnosis
The outcome in the polygenic score calculation was incident AD in the UK Biobank. AD was coded based on the International Classification of Disease (ICD-10) diagnosis code of G30 and READ code F00 (UK Biobank Field ID 131036, and 130836)23. The AD diagnosis in ADNI was based on a clinical diagnosis of expert clinicians following standard research criteria24, while mild cognitive impairment (MCI) was diagnosed based on one of the following: memory complaints, Mini-Mental State Exam (MMSE) (MCI: 24–30, dementia: 20–26), Clinical Dementia Rating score, or Logical Memory II subscale of the Wechsler Memory Scale25,26.
We also used self-reported parental dementia as a secondary outcome to validate the AD PRS in the UK Biobank. Parental dementia status was defined based on all available visits for the reported biological parents and was missing for those who did not complete family history questionnaires: Yes was defined as either parent with a reported diagnosis, Neither as both parents without dementia.
Cognitive function
Participants in the UK Biobank completed their cognitive tests either in assessment centers or during online follow-up27,28,29. We included eight cognitive tests (Fluid intelligence (FI), Matrix pattern completion (MAT), Tower rearranging (TWR), Numeric memory (MEMN), Pairs matching (MEMP), Symbol digit substitution (SDS), Reaction time (RT), and Trail making test B (TMT B)) as the outcomes30 in the association analysis of AD PRS and cognitive function. The scores of some tests were transformed as shown in Supplemental Table 1. For participants with multiple cycles of cognitive tests in the UK Biobank, we only used the tests on the first cycle, and higher cognitive scores meant better cognitive function.
To further validate the AD PRS and cognitive function beyond tests focused on one ___domain, we used general cognitive tests from ADNI. Those cognitive tests included MMSE, the Alzheimer’s Disease Assessment Scale 11 (ADAS 11), Alzheimer’s Disease Assessment Scale 13 (ADAS 13), Montreal Cognitive Assessment (MoCA), and Functional Assessment Questionnaire (FAQ).We also validate the associations of AD PRS with cognitive domains in memory (ADNI_MEM), executive function (ADNI_EF), language (ANDI_LAN), and visuospatial ___domain (ADNI_VS). Methods to calculate these ___domain summary scores can be found elsewhere31,32. In ADNI, participants were administered these tests approximately every 6 months.
Covariates
To control potential confounders in the association of AD PRS and cognitive function, we used age at testing, genotyping array, the first 10 principal components (PCs), smoking status (never, past, current), education years, body mass index (BMI), and apolipoprotein E gene ε4 (APOE4) risk allele (rs429358 and rs7412) dose (0, 1, or 2) as covariates in the UK Biobank. We also used age at testing, sex, education years, first 10 PCs, ever or never smokers (i.e., whether reported a history of cigarette smoking), BMI, and number of APOE4 alleles as covariates in ADNI.
Statistical analysis
We determined the optimal PRS in the training samples composed of unrelated European individuals from the UK Biobank. The Cox proportional hazard model with age, sex, genotyping array, and first 10 PCs adjusted was used to find the optimal AD PRS with maximal C-stats.
Each cognitive measure as well as the polygenic scores were standardized (mean 0 and standard deviation 1). To validate the association of optimal AD PRS and cognitive function internally in the UK Biobank, we linearly regressed the standardized cognitive scores on continuous standardized AD PRS with partially or fully covariates adjustments: Model 1 adjusted for age at cognitive testing, sex, genotyping array, first 10 PCs; Model 2 additionally adjusted for smoking, education in years, and BMI; Model 3 additionally adjusted for the APOE4 risk allele dosages. We also assessed the PRS effects modified by age (less than 60 years vs. aged 60 years and above), APOE4 carrier status, education (high school graduated or not, smoking (ever or never), and obesity (BMI > 30 or not) by adding a multiplicative interaction term into the linear models respectively.
To validate the associations externally, we ran one linear mixed effects model in ADNI with each standardized cognitive score as the dependent variable, continuous standardized AD PRS, age at cognitive testing, sex, education in years, smoking status (never vs. ever), BMI as fixed effects, and participants’ ID as a random effect. To evaluate the effect of APOE on the PRS and cognitive associations we ran an additional mixed effects model with APOE4 alleles as an additional fixed effect. To assess the PRS effects modified by baseline diagnosis and/or APOE4 status we performed analysis stratified by baseline cognitive status (cognitively normal or MCI), and APOE4 status.
Secondary analysis included using categorical AD PRS (PRS lower than 10%, 10–90%, and higher than 90%) as the independent variable to improve clinical interpretations. Logistic regressions were used to assess the association of parental dementia—comparing one parent with dementia to both parents free of dementia, or comparing two parents with dementia to both parents free of dementia—and AD PRS in the UK Biobank. Additionally, the Cox proportional hazard model was employed to examine the PRS associations with incident dementia or incident MCI in the ADNI.
Some cognitive scores (e.g., MMSE, ADAS11, ADAS13) might not normally distributed among cognitively normal or MCI participants in the ADNI, we conducted inverse normal transformation for all the cognitive scores in ADNI and reran the abovementioned linear mixed models as sensitivity analysis.
All the analysis was conducted using R 4.2.1. Specifically, the R package bigsnpr was used to calculate the AD PRS33. We used a two-sided P value < 0.05 and Bonferroni corrections for multiple comparison (UK Biobank significance level = 0.05/8 = 6.25E−03, while ADNI significant level = 0.05/9 = 5.6E−03).
Results
Over 276,000 participants were included in the association analysis of AD PRS and cognitive function in the UK Biobank. The mean age was 57 years (SD = 8 years) and the mean education years was 14.0. Participants from ADNI were generally older with a mean age of 75 years (SD = 6 years) for the cognitively normal group and 72 years (SD = 7 years) for the MCI group. As expected, the percentage of APOE4 carriers was higher in the MCI group (46%) compared with the cognitively normal group (28%). More detailed data on demographic characteristics of the UK Biobank and ADNI participants can be found in Tables 1 and 2, respectively.
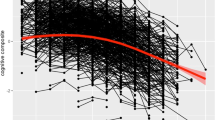
The AD PRS was significantly associated with six of the eight cognitive measures (the exceptions were fluid intelligence and numeric memory) based on the models without APOE4 adjustment. Two cognitive measures (tower rearranging and trail making B) attenuated to null when we additionally adjusted for APOE4 (Table 3). The rest of the measurements (matrix pattern completion (β = − 0.022, P value = 0.003), pairs matching (β = − 0.014, P value = 1.66E−10), symbol digit substitution (β = − 0.017, P value = 1.08E−05), and reaction time (β = − 0.010, P value = 1.19E−06)) were significantly associated with AD PRS even with APOE4 adjustment. The association directions were all negative across varied cognitive measures indicating the higher AD PRS the worse cognitive performance.
Table 4 shows the associations of cognitive function and AD PRS in cognitively normal and MCI participants separately. The MMSE, ADAS 11, ADAS 13 scores were significantly associated with AD PRS in fully adjusted models among cognitively normal individuals. Almost all the comprehensive cognitive scores (MMSE, ADAS 11, ADAS 13, MoCA), functional activity score (FAQ), as well as specific ___domain scores, ADNI memory summary score, ADNI executive function score, and ADNI language summary score) were significantly associated with AD PRS in the partially adjusted models among MCI individuals. Only two cognitive scores (ADAS 11 and ADAS 13) remained nominal significant (i.e., P value < 0.05) when additionally adjusted for APOE4. The association directions also suggested increasing AD PRS was associated with reduced cognitive performance.
The interaction analysis showed potential effects modification by age or APOE4 status but not by education (P interaction ranged from 0.04 to 0.99), smoking status (P interaction ranged from 0.03 to 0.92), or obesity (P interaction ranged from 0.05 to 0.92) in the UK Biobank. Stratification analysis results were presented in Supplemental Tables 2 and Supplemental Table 3. The effect of AD PRS on cognitive function varied by age group (age less than 60, or 60 and above), APOE4 carrier status, and cognitive status (normal or MCI). For example, the effect size of reaction time was larger in the older age group (β = − 0.013, P value = 5.78E−05) than in the younger group (β = − 0.008, P value = 0.004, and P interaction = 0.04). However, we observed stronger associations of AD PRS and symbol digital substitution in the younger UK Biobank population (β = − 0.020, P value = 9.75E−05) than the older ones (β= − 0.016, P value = 9.76E−03, P interaction = 3.42E−14).
The associations of categorical AD PRS (lower than 10%, 10-90%, and 90% and higher) and cognitive function were similar to the continuous AD PRS associations: the mean scores of pair matching and reaction time were lower in the high PRS group, higher in the low PRS group comparing with the middle group (Supplemental Table 4). Other categorical PRS results can be found in Supplemental Tables 5 and Supplemental Table 6.
In secondary analysis, we found positive associations of parental AD with AD PRS in the UK Biobank, the odds of both parents with AD were 2.27 times than those with parents free of AD by 1-SD PRS increment (Supplemental Table 7). For individuals with normal cognition or MCI, the future risk of incident dementia was higher by 25% with 1-SD of PRS increment (Supplemental Table 8). Sensitivity analysis showed consistent association results with or without inverse normalized transformed for those cognitive scores in the ADNI (Supplemental Table 9).
Discussion
Our study validated the associations of AD PRS and cognitive function in both midlife and late-life observational cohorts. Although not all of the cognitive measures were significantly associated with AD PRS, cognitive impairments involving processing speed (such as symbol digit substitution) and short-term memory and attention (such as pairs matching) were associated with the increment of AD PRS in the midlife population (i.e., the UK Biobank). Reduced cognitive performance (e.g., MMSE, ADAS 11, ADAS 13) was also associated with higher AD PRS in the cognitively normal but old population (i.e., the ADNI cognitively normal individuals).
Previous systematic reviews and original investigations illustrated the associations of AD PRS and cognitive measures in populations with relatively small sample sizes22,34. One recent study reported the associations of late-onset AD PRS and fluid intelligence or matrix completion among more than 32,000 individuals from the UK Biobank, but such associations had not been validated by external validation cohorts, nor been assessed through global cognitive function14. Another study from the UK Biobank also suggested that cognitive function diverged in midlife (e.g., aged 45 to 55 years) between higher and lower AD PRS groups13.
Our finding of fluid intelligence and matrix pattern completion and their null associations with AD PRS were not consistent with previous UK Biobank publications14. The PRS was based on the AD GWAS summary statistics derived from varied studies12,35. The PRS improvements in prediction accuracy of “LDpred2” versus the “LDpred” might also contribute to such inconsistencies. We did not find a significant association between AD PRS and numeric memory. Although numeric memory was one of the working memory measures, its subtle changes along with aging and AD PRS increment were comparably smaller than the changes in pairs matching and symbol digit substitution. A prior study also found significant pairs matching and symbol digit differences among high and low PRS groups but not numeric memory13. The associations of comprehensive cognitive measures and AD PRS in the ADNI were similar to a recent ADNI-based study: the correlation of AD PRS and ADAS 13 was stronger than other comprehensive scores such as MMSE and FAQ with and without APOE adjustment36.
The overlapped genes in the AD GWAS and the global cognitive function GWAS could be the biological mechanism behind the relationship between AD PRS and cognitive function37. Global cognitive function GWAS from more than 53,000 individuals identified four risk genes (TOMM40, APOE, ABCG1, and MEF2C), which have also been reported by the AD GWAS37. Other studies also suggested the AD PRS effects on executive function or memory interacted with β-amyloid (Aβ) accumulation: PRS was related to cognitive decline in Aβ + participants but not in Aβ- participants even with APOE4 adjusted38. Another finding in the associations of AD PRS and cognitive function is the role of APOE4 status. Typically, their associations attenuated when further adjusted for APOE4, and more significant PRS (already excluded APOE variants) and cognitive function associations appeared in the APOE4 carriers rather than non-carriers. Another study reported the APOE4 effects on reaction time or memory were not observed nor was an interaction with age or cardiovascular disease39. Characterizing a genetic predisposition to AD solely relying on APOE genotyping has been discouraged40. The modifying effect of APOE4 on the AD PRS could potentially increase the accuracy of risk predictions for AD and early cognitive impairment41. Our study potentially provide evidence that the AD risk stratification models should consider both AD polygenic (e.g., AD PRS) and monogenic (e.g., APOE4) effects.
Although some effects modification by age and APOE4 was found in the UK Biobank, we did not identify the effects modified by other risk factors such as education, BMI, and smoking. Previous study indicated higher educational attainment in early life may attenuate the risk for dementia, particularly among people with high genetic predisposition42. The average education lengths were approximately the same as 14 years across 10% lower or 90% upper PRS group in our data, and made us difficult to find the interaction between PRS and education. Further investigation in other populations or with even larger sample size, or meta-analysis of PRS and cognitive function across multiple cohorts may be warranted.
The clinical significance of AD PRS and its association with cognitive scores is extremely important as these genetic risk factors could be having an impact earlier than what was usually described since we found such associations in middle age population. Identifying individuals who were more likely to have cognitive decline could lead to more targeted preventive measures or treatment strategies depending on their PRS score. Besides the cognitive assessments, genetic information or PRS could serve as one reliable objective evaluation on AD risk stratification and treatment support, potentially in early detection of subtle cognitive impairment.
The strengths of our study include the application of the genome-wide PRS estimation, the validation of our results in an external cohort, and the robustness to multiple covariate adjustments. However, we acknowledge several limitations: the first one is the AD GWAS meta-analysis summary statistics included UK Biobank AD cases, and the AD PRS training in the UK Biobank might have overlapping samples43. Secondly, the diagnosis of AD were different in UK Biobank and ADNI. The AD PRS composed by the UK Biobank might not exactly capture all the genetic predisposition of AD in ADNI, but it could somehow predict incident dementia in ADNI as showed in Supplemental Table 8. Thirdly, residual confounders such as diet, medications, comorbidities of diabetes and cardiovascular diseases might bias the association of AD PRS and cognitive functions, but our results would not significantly change given the UK Biobank was a relatively healthy population and those risk factors effect sizes contributing to cognitive function were trivial29. Lastly, the populations for our study were restricted to European ancestries to minimize the noise of PRS estimates among multiethnic populations. The generalizability was limited and should be further validated in populations of diverse ancestry.
Conclusions
In summary, we validated that cognitive function decrease was associated with higher polygenic AD risk and suggested the usefulness of AD PRS in identifying those who may be at risk for further cognitive decline.
Data availability
AD GWAS summary statistics were downloaded from the European Bioinformatics Institute GWAS Catalog (https://www.ebi.ac.uk/gwas/) under accession no. GCST90027158. The UK Biobank data generated and/or analyzed during current study are not publicly available for privacy reasons, but can be requested through the UK Biobank (https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access). We accessed the data under Application Number 76269. The ADNI whole genome sequencing datasets generated and/or analyzed during the current study are available in the NIAGADS Data Sharing Site (https://dss.niagads.org/sample-sets/snd10002/) with accession number snd10002.
Abbreviations
- AD:
-
Alzheimer’s disease
- PRS:
-
Polygenic risk score
- ADNI:
-
Alzheimer’s Disease Neuroimaging Initiative
- APOE4:
-
Apolipoprotein E gene ε4
- SNVs:
-
Single-nucleotide variants
- GWAS:
-
Genome-wide association studies
- QC:
-
Quality control
- MAF:
-
Minor allele frequency
- BMI:
-
Body mass index
- ICD-10:
-
International Classification of Disease-10
- PCs:
-
Principal components
- FI:
-
Fluid intelligence
- MAT:
-
Matrix pattern completion
- TWR:
-
Tower rearranging
- MEMN:
-
Numeric memory
- MEMP:
-
Pairs matching
- SDS:
-
Symbol digit substitution
- RT:
-
Reaction time
- TMT B:
-
Trail making test B
- MMSE:
-
Mini-Mental State Exam
- ADAS:
-
Alzheimer’s Disease Assessment Scale
- MoCA:
-
Montreal Cognitive Assessment
- FAQ:
-
Functional Assessment Questionnaire
- ADNI_MEM:
-
ADNI memory summary score
- ADNI_EF:
-
ADNI executive function summary score
- ADNI_LAN:
-
ADNI language summary score
- ADNI_VS:
-
ADNI visuospatial summary score
References
Gustavsson, A. et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimers Dement. 19, 658–670 (2023).
Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet (Lond. Engl.) 396, 1204–1222 (2020).
Scheltens, P. et al. Alzheimer’s disease. Lancet (Lond. Engl.) 397, 1577–1590 (2021).
Chaves, M. L. F. et al. Cognitive, functional and behavioral assessment: Alzheimer’s disease. Dement. Neuropsychol. 5, 153–166 (2011).
Riello, M., Rusconi, E. & Treccani, B. The role of brief global cognitive tests and neuropsychological expertise in the detection and Differential diagnosis of Dementia. Front. Aging Neurosci. 13, 648310 (2021).
Parra, M. A. Overcoming barriers in cognitive assessment of Alzheimer’s disease. Dement. Neuropsychol. 8, 95–98 (2014).
Kocagoncu, E. et al. Neurophysiological and brain structural markers of cognitive frailty differ from Alzheimer’s Disease. J. Neurosci. Off J. Soc. Neurosci. 42, 1362–1373 (2022).
Baker, E. et al. What does heritability of Alzheimer’s disease represent? PLoS One 18, e0281440 (2023).
Andrews, S. J. et al. The complex genetic architecture of Alzheimer’s disease: novel insights and future directions. EBioMedicine 90, 104511 (2023).
Escott-Price, V. et al. Common polygenic variation enhances risk prediction for Alzheimer’s disease. Brain 138, 3673–3684 (2015).
Fan, J. et al. The contribution of genetic factors to cognitive impairment and dementia: apolipoprotein E gene, gene interactions, and polygenic risk. Int. J. Mol. Sci. 20, 785 (2019).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Zimmerman, S. C. et al. Association of genetic variants linked to late-onset alzheimer disease with cognitive test performance by midlife. JAMA Netw. open. 5, e225491 (2022).
Tank, R. et al. Association between polygenic risk for Alzheimer’s disease, brain structure and cognitive abilities in UK Biobank. Neuropsychopharmacol. Off Publ Am. Coll. Neuropsychopharmacol. 47, 564–569 (2022).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Privé, F., Arbel, J. & Vilhjálmsson, B. J. LDpred2: better, faster, stronger. Bioinformatics 36, 5424–5431 (2021).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Petersen, R. C. et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 74, 201–209 (2010).
Yang, T. et al. Detecting genetic risk factors for alzheimer’s disease in whole genome sequence data via lasso screening. Proc. IEEE Int. Symp. Biomed. Imaging 2015, 985–989 (2015).
Saykin, A. J. et al. Alzheimer’s disease neuroimaging initiative biomarkers as quantitative phenotypes: genetics core aims, progress, and plans. Alzheimers Dement. 6, 265–273 (2010).
Lin, Y. F. et al. Genetic overlap between vascular pathologies and Alzheimer’s dementia and potential causal mechanisms. Alzheimers Dement. 15, 65–75 (2019).
Wilkinson, T. et al. Identifying dementia outcomes in UK Biobank: a validation study of primary care, hospital admissions and mortality data. Eur. J. Epidemiol. 34, 557–565 (2019).
McKhann, G. et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944 (1984).
Veitch, D. P. et al. Using the Alzheimer’s Disease Neuroimaging Initiative to improve early detection, diagnosis, and treatment of Alzheimer’s disease. Alzheimers Dement. 18, 824–857 (2022).
Hadjichrysanthou, C. et al. The dynamics of biomarkers across the clinical spectrum of Alzheimer’s disease. Alzheimers Res. Ther. 12, 74 (2020).
Hagenaars, S. P. et al. Genetic contributions to trail making test performance in UK Biobank. Mol. Psychiatry 23, 1575–1583 (2018).
Davies, G. et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N = 112 151). Mol. Psychiatry 21, 758–767 (2016).
Hagenaars, S. P. et al. Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N = 112 151) and 24 GWAS consortia. Mol. Psychiatry 21, 1624–1632 (2016).
Lyall, D. M. et al. Correction: cognitive test scores in UK Biobank: data reduction in 480,416 participants and longitudinal stability in 20,346 participants. PLoS One 11, e0156366 (2016).
van Loenhoud, A. C. et al. Cognitive reserve and clinical progression in Alzheimer disease: a paradoxical relationship. Neurology 93, e334–e346 (2019).
Choi, S. E. et al. Development and validation of language and visuospatial composite scores in ADNI. Alzheimer’s Dement. (New York N. Y.) 6, e12072 (2020).
Privé, F., Aschard, H., Ziyatdinov, A. & Blum, M. G. B. Efficient analysis of large-scale genome-wide data with two R packages: bigstatsr and bigsnpr. Bioinformatics 34, 2781–2787 (2018).
Li, Q. et al. Associations of polygenic risk score for late-onset alzheimer’s disease with biomarkers. Front. Aging Neurosci. 14, 849443 (2022).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Suh, E. H. et al. An interpretable Alzheimer’s disease oligogenic risk score informed by neuroimaging biomarkers improves risk prediction and stratification. Front. Aging Neurosci. 15, 1281748 (2023).
Davies, G. et al. Genetic contributions to variation in general cognitive function: a meta-analysis of genome-wide association studies in the CHARGE consortium (N = 53949). Mol. Psychiatry 20, 183–192 (2015).
Ge, T., Sabuncu, M. R., Smoller, J. W., Sperling, R. A. & Mormino, E. C. Dissociable influences of APOE ε4 and polygenic risk of AD dementia on amyloid and cognition. Neurology 90, e1605–e1612 (2018).
Lyall, D. M. et al. Alzheimer disease genetic risk factor APOE e4 and cognitive abilities in 111,739 UK Biobank participants. Age Ageing 45, 511–517 (2016).
Emrani, S., Arain, H. A., DeMarshall, C. & Nuriel, T. APOE4 is associated with cognitive and pathological heterogeneity in patients with Alzheimer’s disease: a systematic review. Alzheimers Res. Ther. 12, 141 (2020).
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C. & Bu, G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518 (2019).
Ma, H. et al. Early-life educational attainment, APOE ε4 alleles, and incident dementia risk in late life. GeroScience 44, 1479–1488 (2022).
Wand, H. et al. Improving reporting standards for polygenic scores in risk prediction studies. Nature 591, 211–219 (2021).
Acknowledgements
This research has been conducted using the UK Biobank Resource under Application Number 76269. We thank the contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible. We extend our appreciation to the participants of the ADNI for their dedicated involvement. This study is indebted to their invaluable contributions, without which it would not have been feasible. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Funding
This work was supported by grants from the National Institute on Aging U01AG058589, P30AG073107, R01AG080670, R01AG083735, and U01AG068221, National Heart, Lung, and Blood Institute R21HL175584, and the American Heart Association (20SFRN35360180). The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Institutes of Health or the US Department of Health and Human Services. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
B.W. and H.L. conceived the study idea. B.W. conducted data analyses, and wrote the first draft of the manuscript. L.B.C., S.H.C., D.B., and A.L.D. reviewed the manuscript critically for important intellectual content. HL supervised the work and made significant contributions to the interpretation of the results and editing of the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All human subjects included in this study provided informed consent. All procedures performed in studies involving human participants were in accordance with the Declaration of Helsinki. The UK Biobank has approval from the North West Multi-centre Research Ethics Committee (MREC) as a Research Tissue Bank (RTB) approval. This research has been conducted using the UK Biobank Resource under Application Number 76269. The ADNI study was approved by the institutional review boards of all the participating institutions, and informed written consent was obtained from all participants at each site. One such institution is the Office for the Protection of Research Subjects at the University of Southern California. More details can be found at adni.loni.usc.edu.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, B., Chibnik, L.B., Choi, S.H. et al. Association of genetic risk of Alzheimer’s disease and cognitive function in two European populations. Sci Rep 15, 6410 (2025). https://doi.org/10.1038/s41598-025-90277-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-90277-9
